





173. Sindroma Down
174. Sindroma Turner
175. Sindroma XXX
176. Sindroma Klinefelter
177. Sindroma XYY
178. Sindroma X Yang Rapuh
179. Sindrom Noonan
180. Sindrom Long QT
Sindroma Down
Sindroma Down
(Trisomi 21, Mongolisme) adalah suatu kelainan kromosom yang
menyebabkan keterbelakangan mental (retardasi mental) dan kelainan
fisik.
Penyakit Trisomi
Penyakit
|
Angka kejadian
|
Kelainan
|
Keterangan
|
Prognosis
|
Trisomi 21
(Sindroma Down |
1 dari 700 bayi baru lahir
|
Kelebihan kromosom 21
|
Perkembangan fisik & mental terganggu, ditemukan berbagai kelainan fisik
|
Biasanya bertahan sampai usia 30-40 tahun
|
Trisomi 18
(Sindroma Edwards) |
1 dari 3.000 bayi baru lahir
|
Kelebihan kromosom 18
|
Kepala kecil, telinga terletak lebih rendah, celah bibir/celah langit-langit, tidak memiliki ibu jari tangan, clubfeet, diantara jari tangan terdapat selaput, kelainan jantung & kelainan saluran kemih-kelamin
|
Jarang bertahan sampai lebih dari beberapa bulan; keterbelakangan mental yg terjadi sangat berat
|
Trisomi 13
(Sindroma Patau) |
1 dari 5.000 bayi baru lahir
|
Kelebihan kromosom 13
|
Kelainan
otak & mata yg berat, celah bibir/celah langit-langit, kelainan
jantung, kelainan saluran kemih-kelamin & kelainan bentuk telinga
|
Yg bertahan hidup sampai lebih dari 1 tahun, kurang dari 20%; keterbelakangan mental yg terjadi sangat berat
|
PENYEBAB
Penyebabnya adalah ekstra tiruan pada kromosom ke 21.
GEJALA
Anak-anak yang menderita sindroma Down memiliki penampilan yang khas:
- Pada saat lahir, ototnya kendur
- Bentuk tulang tengkoraknya asimetris atau ganjil
- Bagian belakang kepalanya mendatar
- Lesi pada iris mata yang disebut bintik Brushfield
- Kepalanya lebih kecil daripada normal (mikrosefalus) dan bentuknya abnormal
- Hidungnya datar, lidahnya menonjol dan matanya sipit ke atas
- Pada sudut mata sebelah dalam terdapat lipatan kulit yang berbentuk bundar (lipatan epikantus)
- Tangannya pendek dan lebar dengan jari-jari tangan yang pendek dan seringkali hanya memiliki 1 garis tangan pada telapak tangannya
- Jari kelingking hanya terdiri dari 2 buku dan melengkung ke dalam
- Telinganya kecil dan terletak lebih rendah
- Diantara jari kaki pertama dan kedua terdapat celah yang cukup lebar
- Gangguan pertumbuhan dan perkembangan (hampir semua penderita sindroma Down tidak pernah mencapai tinggi badan rata-rata orang dewasa)
- Keterbelakangan mental.


Pada bayi yang menderita sindroma Down sering ditemukan kelainan jantung bawaan. Kematian dini seringkali terjadi akibat kelainan jantung.
Kelainan saluran pencernaan, seperti atresia esofagus (penyumbatan kerongkongan) dan atresia duodenum (penyumbatan usus 12 jari), juga sering ditemukan.
Mereka juga memiliki resiko tinggi menderita leukemia limfositik akut.
DIAGNOSA
Diagnosis sindroma Down dapat ditegakkan ketika bayi masih berada dalam kandungan dan tes penyaringan biasanya dilakukan pada wanita hamil yang berusia diatas 35 tahun.
Kadar alfa-fetoprotein yang rendah di dalam darah ibu menunjukkan resiko tinggi terjadinya sindroma Down pada janin yang dikandungnya.
Dengan pemeriksaan USG bisa diketahui adanya kelainan fisik pada janin.
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik.
Dengan bantuan stetoskop akan terdengar murmur (bunyi jantung tambahan).
Pemeriksaan yang biasa dilakukan:
- Analisa kromosom (pada 94% kasus menunjukkan adanya 3 tiruan dari kromosom ke 21)
- Rontgen dada (untuk menunjukkan adanya kelainan jantung)
- Ekokardiogram
- EKG
- Rontgen saluran pencernaan.
PENGOBATAN
Tidak ada pengobatan khusus untuk sindroma Down. Pendidikan dan pelatihan khusus bisa dilakukan di sekolah luar biasa.
Kelainan jantung tertentu mungkin harus diperbaiki melalui pembedahan.
Gangguan pendengaran dan penglihatan diatasi sebagaimana mestinya.
PROGNOSIS
Anak-anak dengan sindroma Down memiliki resiko tinggi untuk menderita kelainan jantung dan leukemia. Jika terdapat kedua penyakit tersebut, maka angka harapan hidupnya berkurang; jika kedua penyakit tersebut tidak ditemukan, maka anak bisa bertahan sampai dewasa.
Beberapa penderita sindroma Down mengalami hal-hal berikut:
- Gangguan tiroid
- Gangguan pendengaran akibat infeksi telinga berulang dan otitis serosa
- Gangguan penglihatan karena adanya perubahan pada lensa dan kornea
- Pada usia 30 tahun menderita demensia (berupa hilang ingatan, penurunan kecerdasan dan perubahan kepribadian).
Bisa terjadi kematian dini, meskipun banyak juga penderita yang berumur panjang.
PENCEGAHAN
Pada keluarga yang memiliki riwayat sindroma Down dianjurkan untuk menjalani konsultasi genetik.
Sindroma Down bisa diketahui pada kehamilan awal dengan melakukan pemeriksaan kromosom terhadap cairan ketuban atau vili korion.
Resiko terjadinya sindroma Down ditemukan pada:
- keluarga yang pernah memiliki anak yang menderita sindroma Down
- ibu hamil yang berusia diatas 40 tahun.
Sindroma Turner
Sindroma Turner
(Disgenesis Gonad) adalah suatu keadaan pada anak perempuan, dimana
salah satu dari kromosom Xnya hilang sebagian atau hilang seluruhnya.
PENYEBAB
Sindroma Turner biasanya disebabkan oleh hilangnya kromosom X.
Kelainan ini ditemukan pada 1 diantara 3.000 bayi baru lahir.
Sindroma Turner bukan merupakan penyakit keturunan. Tetapi kadang salah satu orang tua membawa kromosom yang telah mengalami penyusunan ulang, yang bisa menyebabkan terjadinya sindroma ini.
GEJALA
Gejalanya adalah:

DIAGNOSA
Sindroma Turner mungkin terdiagnosis pada saat bayi lahir karena adanya kelainan-kelainan tersebut diatas atau mungkin juga baru terdiagnosis pada masa pubertas karena adanya menstruasi dan perkembangan ciri seksual sekunder yang tertunda.
Pemeriksaan fisik menunjukkan payudara dan alat kelamin yang kurang berkembang, webbed neck, tubuh yang pendek dan kelainan bentuk dada.
Pemeriksaan yang biasa dilakukan:
- Kariotip
- USG
- Pemeriksaan ginekologis
- Peningkatan kadar LH dan FSH.
PENGOBATAN
Pengobatannya bersifat suportif.
Agar tinggi badannya bertambah, bisa diberikan hormon pertumbuhan.
Terapi estrogen dimulai pada usia 12-13 tahun untuk merangsang pertumbuhan ciri seksual sekunder sehingga penderita akan memiliki penampilan yang lebih normal pada masa dewasa nanti. Tetapi terapi estrogen tidak dapat mengatasi kemandulan.
Untuk mencegah kekeringan, rasa gatal dan nyeri selama melakukan hubungan seksual, bisa digunakan pelumas vagina.
Untuk memperbaiki kelainan jantung kadang perlu dilakukan pembedahan.
PENYEBAB
Sindroma Turner biasanya disebabkan oleh hilangnya kromosom X.
Kelainan ini ditemukan pada 1 diantara 3.000 bayi baru lahir.
Sindroma Turner bukan merupakan penyakit keturunan. Tetapi kadang salah satu orang tua membawa kromosom yang telah mengalami penyusunan ulang, yang bisa menyebabkan terjadinya sindroma ini.
GEJALA
Gejalanya adalah:
- Tubuh pendek
- Webbed neck (kulit diantara leher dan bahunya menyatu, seperti selaput)
- Garis rambut yang pendek pada leher bagian belakangnya
- Kelopak matanya turun
- Pembengkakan pada punggung tangan dan puncak kakinya (limfedema)
- Pada leher bagian belakang seringkali ditemukan pembengkakan atau lipatan kulit yang longgar
- Jari manis dan jari-jari kakinya pendek, kukunya tidak terbentuk dengan baik
- Perkembangan tulang abnormal (misalnya dada berbentuk seperti tameng, lebar dan datar, dengan jarak yang lebar diantara kedua puting susunya)
- Pada kulitnya terdapat banyak tahi lalat berwarna gelap
- Perkembangan seksual sekunder pada masa pubertas tidak terjadi atau mengalami keterbelakangan (rambut kemaluan yang jarang dan tipis, payudara kecil)
- Kemandulan, karena ovarium (sel indung telur) biasanya mengandung sel-sel telur yang tidak berkembang
- Pembentukan air mata berkurang
- Amenore (tidak mengalami menstruasi)
- Simian crease (pada telapak tangan hanya terdapat satu garis tangan)
- Kelembaban vagina tidak ada sehingga hubungan seksual menimbulkan rasa nyeri
- Koartasio aorta (penyempitan aorta), yang bisa menyebabkan tekanan darah tinggi
- Sering ditemukan kelainan ginjal dan pembengkakan pada pembuluh darah (hemangioma)
- Kadang kelainan pembuluh darah pada usus menyebabkan terjadinya perdarahan
- Kadang terjadi keterbelakangan mental.
DIAGNOSA
Sindroma Turner mungkin terdiagnosis pada saat bayi lahir karena adanya kelainan-kelainan tersebut diatas atau mungkin juga baru terdiagnosis pada masa pubertas karena adanya menstruasi dan perkembangan ciri seksual sekunder yang tertunda.
Pemeriksaan fisik menunjukkan payudara dan alat kelamin yang kurang berkembang, webbed neck, tubuh yang pendek dan kelainan bentuk dada.
Pemeriksaan yang biasa dilakukan:
- Kariotip
- USG
- Pemeriksaan ginekologis
- Peningkatan kadar LH dan FSH.
PENGOBATAN
Pengobatannya bersifat suportif.
Agar tinggi badannya bertambah, bisa diberikan hormon pertumbuhan.
Terapi estrogen dimulai pada usia 12-13 tahun untuk merangsang pertumbuhan ciri seksual sekunder sehingga penderita akan memiliki penampilan yang lebih normal pada masa dewasa nanti. Tetapi terapi estrogen tidak dapat mengatasi kemandulan.
Untuk mencegah kekeringan, rasa gatal dan nyeri selama melakukan hubungan seksual, bisa digunakan pelumas vagina.
Untuk memperbaiki kelainan jantung kadang perlu dilakukan pembedahan.
Sindroma XXX
Manusia memiliki 46 kromosom, 2 diantaranya adalah kromosom seks yang menunjukkan jenis kelaminnya. Dalam keadaan normal, wanita memiliki 2 kromosom X (digambarkan sebagai 46,XX), sedangkan pria memiliki 1 kromosom X dan 1 kromosom Y (digambarkan sebagai 46,XY).
Pada sindroma XXX, di setiap sel-sel tubuh wanita terdapat 3 kromosom X (digambarkan sebagai 47,XXX).
Sekitar 1 diantara 1.000 bayi perempuan yang tampaknya normal, memiliki kelainan ini.
Gejalanya sangat bervariasi. Beberapa penderita tidak menunjukkan gejala atau gejalanya sangat sedikit, sedangkan penderita lainnya memiliki gambaran yang lebih berat.
Bayi seringkali tenang dan tidak aktif, Mereka mengalami perkembangan fungsi motorik, berbicara dan pematangan yang tertunda.
Penundaan perkembangan ini sebaiknya diatasi dengan memberikan rangsangan fungsi mental, sosial dan motoriknya, baik di rumah maupun di klinik khusus.
Jika terjadi gangguan perkembangan berbicara, perlu dilakukan terapi bicara.
Anak perempuan dengan 3 kromosom X cenderung memiliki tingkat kecerdasan yang lebih rendah dibandingkan dengan saudara laki-laki dan saudara perempuannya yang normal .
Kadang sindroma ini menyebabkan kemandulan, meskipun beberapa penderita bisa melahirkan anak yang memiliki kromosom dan fisik yang normal. Beberapa penderita mengalami menopause dini.
Sindroma Klinefelter
Pada sindroma Klinefelter, bayi laki-laki terlahir dengan kelebihan 1 kromosom X (digambarkan sebagai 47,XXY).
Pria dan wanita biasanya memiliki 2 kromosom seks. Wanita mendapatkan 2 kromosom X, 1 dari ibu, 1 dari ayah. Pria mendapatkan 1 kromosom X dari ibu dan 1 kromosom Y dari ayah.
Pria dengan sindroma Klinefelter biasanya memiliki kelebihan kromosom X sehingga mereka memiliki 3 kromosom seks, yaitu 2 kromosom X dan 1 kromosom Y.
Sindroma ini ditemukan pada 1 diantara 700 bayi baru lahir.
Jumlah kromosom pada pria & wanita

PENYEBAB
Penyebabnya tidak diketahui.
GEJALA
Gejalanya berupa:
- Pembesaran buah dada (ginekomastia)
- Rambut wajah dan rambut tubuh yang jarang dan tipis
- Bentuk tubuhnya lebih bundar
- Testis (buah zakar) kecil dan tidak mampu menghasilkan sperma
- Cenderung lebih mudah mengalami obesitas (kegemukan)
- Cenderung memiliki tubuh yang lebih tinggi.
Biasanya tidak terjadi keterbelakangan mental, tetapi banyak yang mengalami gangguan berbahasa.
Pada masa kanak-kanak, mereka seringkali mengalami keterlambatan dalam berbicara dan mungkin mengalami kesulitan dalam belajar membaca dan menulis.
Jika tidak diobati, gangguan berbahasa ini bisa menyebabkan kegagalan di sekolah dan mengurangi rasa percaya diri. Pengobatan terhadap gangguan berbahasa sebaiknya dilakukan pada awal masa kanak-kanak.

DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala, hasil pemeriksaan fisik dan hasil analisa kromosom (kariotip).
Diagnosis bisa ditegakkan pada berbagai keadaan:
# Bayi masih berada dalam kandungan.
Diagnosis ditegakkan melalui pemeriksaan amniosintesis (analisa cairan ketuban).
Prosedur ini tidak dilakukan secara rutin, tetapi hanya dilakukan jika terdapat riwayat keluarga dengan kelainan genetik atau jika usia ibu lebih dari 35 tahun.
# Pada awal masa kanak-kanak.
Diduga suatu sindroma Klinefelter jika seorang anak laki-laki terlambat berbicara dan mengalami kesulitan dalam membaca serta menulis. Anak laki-laki dengan XXY tampak lebih tinggi dan kurus, serta pasif dan pemalu.
# Remaja.
Remaja laki-laki merasa malu ketika menyadari bahwa payudaranya agak membesar, karena itu mereka berobat ke dokter.
# Dewasa.
Diagnosis biasanya merupakan akibat dari adanya kemandulan. Pada pemeriksaan fisik, testis tampak lebih kecil. Untuk memperkuat diagnosis sindroma ini, dilakukan pemeriksaan kadar hormon gonadotropin.
PENGOBATAN
Efek yang utama dari sindroma Klinefelter adalah pada fungsi testis. Testis menghasilkan hormon pria testosteron dan jumlah hormon ini pada penderita sindroma Klinefelter menurun.
Pada saat penderita berusia 10-12 tahun, perlu dilakukan pengukuran testosteron dalam darahnya secara periodik (misalnya setiap tahun). Jika kadarnya rendah (sehingga tidak terjadi perubahan seksual yang seharusnya dialami setiap anak laki-laki pada masa pubertas) atau jika timbul gejala yang disebabkan oleh gangguan metabolisme hormon, maka dilakukan pengobatan dengan pemberian hormon testosteron.
Yang paling sering digunakan adalah depotestosteron, yang merupakan hormon testosteron sintetis, disuntikkan 1 kali/bulan. Sejalan dengan pertambahan umur penderita, secara bertahap dosisnya perlu ditingkatkan dan diberikan lebih sering.
Hasil dari pengobatan adalah perkembangan fisik dan seksual yang normal, yaitu berupa pertumbuhan rambut kemaluan, penambahan ukuran penis dan skrotum (kantung zakar), pertumbuhan janggut, suara menjadi lebih dalam serta otot lebih berisi dan lebih kuat.
Keuntungan lain yang diperoleh dari terapi testosteron adalah:
- Pikiran lebih jernih
- Lebih bertenaga
- Tremor tangan berkurang
- Pengendalian diri yang lebih baik
- Dorongan seksual lebih besar
- Lebih mudah menyesuaikan diri di sekolah dan tempat bekerja
- Lebih percaya diri.
Pria dewasa mampu menjalani fungsi seksual yang normal (ereksi dan ejakulasi), tetapi tidak mampu menghasilkan sperma dalam jumlah yang normal.
Pria dan wanita biasanya memiliki 2 kromosom seks. Wanita mendapatkan 2 kromosom X, 1 dari ibu, 1 dari ayah. Pria mendapatkan 1 kromosom X dari ibu dan 1 kromosom Y dari ayah.
Pria dengan sindroma Klinefelter biasanya memiliki kelebihan kromosom X sehingga mereka memiliki 3 kromosom seks, yaitu 2 kromosom X dan 1 kromosom Y.
Sindroma ini ditemukan pada 1 diantara 700 bayi baru lahir.
Jumlah kromosom pada pria & wanita
PENYEBAB
Penyebabnya tidak diketahui.
GEJALA
{kind=link}
Gejalanya berupa:
- Pembesaran buah dada (ginekomastia)
- Rambut wajah dan rambut tubuh yang jarang dan tipis
- Bentuk tubuhnya lebih bundar
- Testis (buah zakar) kecil dan tidak mampu menghasilkan sperma
- Cenderung lebih mudah mengalami obesitas (kegemukan)
- Cenderung memiliki tubuh yang lebih tinggi.
Biasanya tidak terjadi keterbelakangan mental, tetapi banyak yang mengalami gangguan berbahasa.
Pada masa kanak-kanak, mereka seringkali mengalami keterlambatan dalam berbicara dan mungkin mengalami kesulitan dalam belajar membaca dan menulis.
Jika tidak diobati, gangguan berbahasa ini bisa menyebabkan kegagalan di sekolah dan mengurangi rasa percaya diri. Pengobatan terhadap gangguan berbahasa sebaiknya dilakukan pada awal masa kanak-kanak.
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala, hasil pemeriksaan fisik dan hasil analisa kromosom (kariotip).
Diagnosis bisa ditegakkan pada berbagai keadaan:
# Bayi masih berada dalam kandungan.
Diagnosis ditegakkan melalui pemeriksaan amniosintesis (analisa cairan ketuban).
Prosedur ini tidak dilakukan secara rutin, tetapi hanya dilakukan jika terdapat riwayat keluarga dengan kelainan genetik atau jika usia ibu lebih dari 35 tahun.
# Pada awal masa kanak-kanak.
Diduga suatu sindroma Klinefelter jika seorang anak laki-laki terlambat berbicara dan mengalami kesulitan dalam membaca serta menulis. Anak laki-laki dengan XXY tampak lebih tinggi dan kurus, serta pasif dan pemalu.
# Remaja.
Remaja laki-laki merasa malu ketika menyadari bahwa payudaranya agak membesar, karena itu mereka berobat ke dokter.
# Dewasa.
Diagnosis biasanya merupakan akibat dari adanya kemandulan. Pada pemeriksaan fisik, testis tampak lebih kecil. Untuk memperkuat diagnosis sindroma ini, dilakukan pemeriksaan kadar hormon gonadotropin.
PENGOBATAN
Efek yang utama dari sindroma Klinefelter adalah pada fungsi testis. Testis menghasilkan hormon pria testosteron dan jumlah hormon ini pada penderita sindroma Klinefelter menurun.
Pada saat penderita berusia 10-12 tahun, perlu dilakukan pengukuran testosteron dalam darahnya secara periodik (misalnya setiap tahun). Jika kadarnya rendah (sehingga tidak terjadi perubahan seksual yang seharusnya dialami setiap anak laki-laki pada masa pubertas) atau jika timbul gejala yang disebabkan oleh gangguan metabolisme hormon, maka dilakukan pengobatan dengan pemberian hormon testosteron.
Yang paling sering digunakan adalah depotestosteron, yang merupakan hormon testosteron sintetis, disuntikkan 1 kali/bulan. Sejalan dengan pertambahan umur penderita, secara bertahap dosisnya perlu ditingkatkan dan diberikan lebih sering.
Hasil dari pengobatan adalah perkembangan fisik dan seksual yang normal, yaitu berupa pertumbuhan rambut kemaluan, penambahan ukuran penis dan skrotum (kantung zakar), pertumbuhan janggut, suara menjadi lebih dalam serta otot lebih berisi dan lebih kuat.
Keuntungan lain yang diperoleh dari terapi testosteron adalah:
- Pikiran lebih jernih
- Lebih bertenaga
- Tremor tangan berkurang
- Pengendalian diri yang lebih baik
- Dorongan seksual lebih besar
- Lebih mudah menyesuaikan diri di sekolah dan tempat bekerja
- Lebih percaya diri.
Pria dewasa mampu menjalani fungsi seksual yang normal (ereksi dan ejakulasi), tetapi tidak mampu menghasilkan sperma dalam jumlah yang normal.
Sindroma XYY
Pada sindroma XYY, seorang bayi laki-laki terlahir dengan kelebihan kromosom Y.
Pria biasanya hanya memiliki 1 kromosom X dan 1 kromosom Y, digambarkan sebagai 46,XY.
Pria dengan sindroma XYY memiliki 2 kromosom Y dan digambarkan sebagai 47,XYY.
Kelainan ini ditemukan pada 1 diantara 1.000 pria.
Jumlah kromosom pada pria & wanita

PENYEBAB
Penyebab dari penyimpangan kromosom yang menyebabkan terbentuknya sindroma XYY tidak diketahui.
GEJALA
Pada saat lahir, bayi biasanya tampak normal, lahir dengan berat dan panjang badan yang normal, tanpa kelainan fisik dan organ seksualnya normal.
Pada awal masa kanak-kanak, penderita memiliki kecepatan pertumbuhan yang pesat, rata-rata mereka memiliki tinggi badan 7 cm diatas normal.
Postur tubuhnya normal, tetapi berat badannya relatif lebih rendah jika dibandingkan terhadap tinggi badannya.
Pada masa kanak-kanak, mereka lebih aktif dan cenderung mengalami penundaan kematangan mental, meskipun fisiknya berkembang secara normal dan tingkat kecerdasannya berada dalam kisaran normal.
Di sekolah, mereka cenderung mengalami masalah belajar.
Aktivitas yang tinggi dan gangguan belajar akan menimbulkan masalah di sekolah sehingga perlu diberikan pendidikan ekstra.
Perkembangan seksual fisiknya normal, dimana organ seksual dan ciri seksual sekundernya berkembang secara normal. Pubertas terjadi pada waktunya.
Pria XYY tidak mandul, mereka memilki testis yang berukuran normal serta memiliki potensi dan gairah seksual yang normal.

DIAGNOSA
Diagnosis ditegakkan berdasarkan hasil pemeriksaan analisa kromosom.
PENGOBATAN
Anak laki-laki dengan sindroma XYY seringkali secara fisik lebih aktif daripada saudara kandungnya dan jika aktivitas ini ditanggapi dan disalurkan dengan baik, biasanya tidak akan menimbulkan masalah.
Mereka cenderung mengalami keterlambatan dalam kematangan emosi dan cenderung mengalami kesulitan belajar di sekolah sehingga perlu dirangsang secara dini dan adekuat.
Pria XYY memiliki keadaan hormon seks yang normal dan tidak perlu menjalani terapi hormonal.
Anak laki-laki XYY yang tumbuh di dalam lingkungan yang baik, dengan cinta, dukungan dan rangsangan yang mereka perlukan, tidak akan mengalami kelainan jiwa.
Pria XYY yang tumbuh dalam lingkungan yang jelek, tanpa cinta, rangsangan dan dukungan, memiliki resiko mengalami kelainan jiwa dan gangguan dalam bersosialisasi; tetapi mereka tidak memiliki resiko menderita skizofrenia, kelainan manik-depresif maupun kelainan jiwa yang serius lainnya. Mereka bisa dibantu melalui penyuluhan dan pengobatan psikolog-psikiater.
Pria biasanya hanya memiliki 1 kromosom X dan 1 kromosom Y, digambarkan sebagai 46,XY.
Pria dengan sindroma XYY memiliki 2 kromosom Y dan digambarkan sebagai 47,XYY.
Kelainan ini ditemukan pada 1 diantara 1.000 pria.
Jumlah kromosom pada pria & wanita
PENYEBAB
Penyebab dari penyimpangan kromosom yang menyebabkan terbentuknya sindroma XYY tidak diketahui.
GEJALA
Pada saat lahir, bayi biasanya tampak normal, lahir dengan berat dan panjang badan yang normal, tanpa kelainan fisik dan organ seksualnya normal.
Pada awal masa kanak-kanak, penderita memiliki kecepatan pertumbuhan yang pesat, rata-rata mereka memiliki tinggi badan 7 cm diatas normal.
Postur tubuhnya normal, tetapi berat badannya relatif lebih rendah jika dibandingkan terhadap tinggi badannya.
Pada masa kanak-kanak, mereka lebih aktif dan cenderung mengalami penundaan kematangan mental, meskipun fisiknya berkembang secara normal dan tingkat kecerdasannya berada dalam kisaran normal.
Di sekolah, mereka cenderung mengalami masalah belajar.
Aktivitas yang tinggi dan gangguan belajar akan menimbulkan masalah di sekolah sehingga perlu diberikan pendidikan ekstra.
Perkembangan seksual fisiknya normal, dimana organ seksual dan ciri seksual sekundernya berkembang secara normal. Pubertas terjadi pada waktunya.
Pria XYY tidak mandul, mereka memilki testis yang berukuran normal serta memiliki potensi dan gairah seksual yang normal.
DIAGNOSA
Diagnosis ditegakkan berdasarkan hasil pemeriksaan analisa kromosom.
PENGOBATAN
Anak laki-laki dengan sindroma XYY seringkali secara fisik lebih aktif daripada saudara kandungnya dan jika aktivitas ini ditanggapi dan disalurkan dengan baik, biasanya tidak akan menimbulkan masalah.
Mereka cenderung mengalami keterlambatan dalam kematangan emosi dan cenderung mengalami kesulitan belajar di sekolah sehingga perlu dirangsang secara dini dan adekuat.
Pria XYY memiliki keadaan hormon seks yang normal dan tidak perlu menjalani terapi hormonal.
Anak laki-laki XYY yang tumbuh di dalam lingkungan yang baik, dengan cinta, dukungan dan rangsangan yang mereka perlukan, tidak akan mengalami kelainan jiwa.
Pria XYY yang tumbuh dalam lingkungan yang jelek, tanpa cinta, rangsangan dan dukungan, memiliki resiko mengalami kelainan jiwa dan gangguan dalam bersosialisasi; tetapi mereka tidak memiliki resiko menderita skizofrenia, kelainan manik-depresif maupun kelainan jiwa yang serius lainnya. Mereka bisa dibantu melalui penyuluhan dan pengobatan psikolog-psikiater.
Sindroma X Yang Rapuh
PENYEBAB
Sindroma ini terjadi pada sepertiga dari semua keterbelakangan mental yang berkaitan dengan kromosom X pada pria dan pada wanita sebesar sepersepuluhnya.
Keadaan ini terjadi pada 1 diantara 2.000 pria dan 1 diantara 4.000 wanita.
Sindroma X yang rapuh diturunkan dengan pola resesif X-linked, artinya anak laki-laki lebih mungkin menderita sindroma ini dan gen yang bermutasi dibawa oleh kedua orangtuanya.
GEJALA
Gejalanya berupa:
- Keterbelakangan mental
- Cenderung menghindari kontak mata
- Perilaku hiperaktif
- Dahi dan telinga lebar, rahang yang menonjol
- Testis (buah zakar) yang besar.
DIAGNOSA
Untuk mendiangosis sindroma ini, dilakukan analisa kromosom dan pemeriksaan genetik khusus PCR.
Salah satu tanda dari sindroma X yang rapuh adalah bayi cenderung memiliki lingkar kepala yang besar atau testis yang sangat besar.
Ciri khas dari sindroma ini adalah keterbelakangan mental.
PENGOBATAN
Tidak ada pengobatan khusus untuk sindroma X yang rapuh.
Berbagai usaha ditujukan kepada pelatihan dan pendidikan sehingga anak bisa berfungsi senormal mungkin.
PENCEGAHAN
Jika di dalam keluarga ada riwayat sindroma X yang rapuh, dianjurkan untuk menjalani konsultasi genetik untuk mengetahui resiko terjadinya sindroma yang sama pada keturunannya.
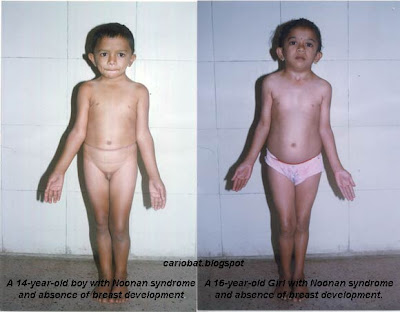
Sindrom Noonan
Sindrom Noonan
adalah kerusakan genetik yang menyebabkan sejumlah kelainan fisik,
biasanya termasuk perawakan pendek, kerusakan jantung, dan penampilan
yang tidak normal.
Sindrom Noonan bisa diwarisi atau bisa terjadi tanpa diprediksi pada anak yang orangtuanya memiliki gen normal. meskipun anak dengan sindrom tersebut mempunyai struktur kromosom yang normal, mereka memiliki banyak sifat khas pada sindrom Turner. Dulu, sindrom Noonan disebut ‘sindrom Turner lelaki’. Anak laki-laki atau anak perempuan bisa terkena. Gen bertanggung jawab untuk sindrom Noonan sudah dilokalisasikan ke kromosom 12.
PENYEBAB
Diperkirakan sindrom Noonan terjadi satu dari 1.000 sampai 2.500 kelahiran. Penyebabnya adalah mutasi pada gen tertentu. Saat ini, para ilmuwan tahu dari empat gen yang dapat menyebabkan sindrom Noonan. Mutasi dapat diwariskan dari orangtua yang membawa gen cacat (autosomal dominan), atau bisa berkembang karena mutasi baru pada anak-anak yang tidak memiliki kecenderungan genetik untuk penyakit ini. Anak-anak yang memiliki satu orangtua dengan sindrom Noonan memiliki peluang 50 persen mengalami gangguan ini.
Keempat gen yang diketahui bahwa dapat bermutasi dan menyebabkan sindrom Noonan adalah:
- PTPN11 gen. Sekitar separuh orang-orang dengan sindrom Noonan mendapatkan gangguan karena mutasi gen bernama PTPN11.
- SOS1 gen. Sebanyak 20 persen orang dengan sindrom Noonan memiliki kondisi karena cacat pada gen ini.
- RAF1 gen. gen ini bertanggung jawab ampai 15 persen dari semua kasus sindrom Noonan.
- Gen Kras. Sekitar 5 sampai 10 persen orang mendapatkan gangguan karena mutasi pada gen Kras. Orang dengan jenis mutasi ini mungkin memiliki bentuk yang lebih parah dari sindrom Noonan.
Cacat dalam gen menyebabkan mereka memproduksi protein yang terus aktif. Karena gen ini berperan dalam pembentukan jaringan di seluruh tubuh, aktivasi konstan protein ini mengganggu proses normal dari pertumbuhan sel dan pembelahan.
Pada beberapa orang, tidak ada mutasi pada gen di atas, yang artinya bisa gen lainnya, masih belum ditemukan gen lain yang menyebabkan sindrom Noonan. Dalam kasus yang jarang terjadi, orang-orang yang memiliki karakteristik sindrom Noonan juga memiliki kondisi lain yang disebut neurofibromatosis 1. Orang dengan kedua kondisi memiliki mutasi pada gen neurofibromin 1, menghasilkan protein yang membantu menjaga sel-sel untuk tidak tumbuh terlalu cepat atau dengan cara yang tidak terkendali.
GEJALA
Gejalanya bisa termasuk anyaman pada leher, telinga letaknya rendah, kelopak mata sayu, perawakan pendek, jari keempat yang memendek (ring), langit-langit mulut melengkung tinggi, dan kelainan jantung dan pembuluh darah. kecerdasan bisa terganggu. Yang paling mempengaruhi orang adalah pendek. Anak laki-laki bisa memiliki testes yang tidak berkembang atau tidak turun. Pada anak perempuan, indung telur kemungkinan tidak aktif atau berhenti bekerja. Pubertas kemungkinan terlambat, dan kemandulan bisa terjadi.
DIAGNOSA
Diagnosis sindrom Noonan biasanya dilakukan setelah dokter mengamati beberapa dari tanda-tanda dan gejala kunci penyakit ini, tetapi ini bisa sulit karena beberapa fitur yang terkait dengan gangguan ini adalah samar hingga sulit untuk mengidentifikasi. Sering kali, sindrom Noonan tidak terdiagnosa sampai dewasa, setelah seseorang memiliki anak yang lebih jelas dipengaruhi oleh kondisi. Pengujian molekul genetik dapat digunakan untuk membuat diagnosis yang pasti.
Jika ada bukti masalah jantung, dokter mungkin akan merekomendasikan untuk menjalani elektrokardiogram (EKG) dan ekokardiogram untuk menilai tipe dan tingkat keparahan kondisi. EKG ini dilakukan dengan menempatkan elektroda di dada Anda. Sebuah EKG terlihat pada impuls listrik yang berjalan melalui jantung Anda untuk menilai masalah. Suatu ekokardiogram menggunakan gelombang suara untuk menciptakan sebuah gambar bergerak dari kerja jantungi Anda agar dokter Anda dapat melihat di mana ada masalah. Tes-tes ini biasanya dilakukan oleh dokter spesialis jantung.
PENGOBATAN
Pertumbuhan kemungkinan meningkat oleh pengobatan dengan hormon pertumbuhan. Setelah pertumbuhan tercukupi, pengobatan testosteron bisa membantu anak laki-laki yang mempunya testes tidak berkembang. Testosteron merangsang perkembangan penampilan lebih maskulin.
PENCEGAHAN
Jika Anda memiliki riwayat keluarga memiliki sindrom Noonan, konsultasi dengan dokter dan jalani konseling genetik sebelum memiliki anak. Namun, karena banyak kasus penyakit ini terjadi secara spontan, tidak ada cara yang diketahui untuk mencegahnya.
Jika sindrom Noonan terdeteksi lebh awal, mungkin dengan perawatan berkelanjutan dan komprehensif dapat mengurangi beberapa komplikasinya, seperti penyakit jantung.
Sindrom Long QT
Sindrom long QT adalah kelainan pada
sistem elektrikal jantung, yang bisa menyebabkan hilangnya kesadaran
atau kematian tiba-tiba.
PENYEBAB
Sindrom long QT bisa diderita 1 dari 7.000 orang. Di Amerika Serikat, hal ini bisa menyebabkan kematian tiba-tiba dalam 3.000 sampai 4.000 anak dan orang dewasa muda setiap tahunnya. Pada anak, gangguan ini biasanya berhubungan dengan kelainan genetik. Seseorang dengan gangguan tersebut bisa memiliki anggota keluarga yang meninggal secara tiba-tiba dan tidak dapat dijelaskan. Pada kebanyakan orang dewasa, sindrom long QT disebabkan oleh penggunaan obat atau sebuah gangguan.
GEJALA
Orang yang mengalami sindrom long QT mempengaruhi terbentuknya detak jantung yang cepat, yang seringkali terjadi selama kegiatan fisik atau kegembiraan emosional. Ketika detak jantung terlalu cepat, otak bisa tidak menerima cukup darah. Akibatnya adalah kehilangan kesadaran. Beberapa orang dengan sindrom long QT juga terlahir tuli. Tetapi sekitar sepertiga orang tidak mengalami gejala-gejala. Sindrom long QT bisa menyebabkan kematian tiba-tiba pada usia muda.
DIAGNOSA
Dokter bisa menganjurkan electrocardiogfrafi (ECG) untuk anak atau orang dewasa muda yang mengalami hilangnya kesadaran secara tiba-tiba dan tidak dapat dijelaskan. Prosedur tersebut kemungkinan dilakukan dengan orang tersebut ketika istirahat atau setelah menerima obat-obatan secara infus atau orang tersebut kemungkinan diminta untuk berjalan di atas treadmill atau pedal sebuah sepeda latihan dalam prosedur yang disebut tes latihan stress.
PENGOBATAN
Beta-blocker efektif untuk kebanyakan anak dan orang dewasa. Beberapa orang dewasa bisa menerima manfaat dari mexiletine, sebuah obat antiaritmia. Untuk anak dan orang dewasa yang tidak bereaksi terhadap obat-obatan, sebuah pacemaker atau kombinasi pacemaker-internal defibrillator kemungkinan dicoba. Internal defibrillator bisa mengejutkan jantung, menghidupkan kembali orang tersebut, kapan saja jantung tersebut membentuk kelainan ritme yang mematikan. Kadangkala, sebagai alternatif, saraf pada leher dipotong dalam prosedur yang disebut cervicothoracic sympathectomy. Pemotongan saraf ini bisa membantu mencegah detak jantung cepat yang menyebabkan kematian tiba-tiba.
PENYEBAB
Sindrom long QT bisa diderita 1 dari 7.000 orang. Di Amerika Serikat, hal ini bisa menyebabkan kematian tiba-tiba dalam 3.000 sampai 4.000 anak dan orang dewasa muda setiap tahunnya. Pada anak, gangguan ini biasanya berhubungan dengan kelainan genetik. Seseorang dengan gangguan tersebut bisa memiliki anggota keluarga yang meninggal secara tiba-tiba dan tidak dapat dijelaskan. Pada kebanyakan orang dewasa, sindrom long QT disebabkan oleh penggunaan obat atau sebuah gangguan.
GEJALA
Orang yang mengalami sindrom long QT mempengaruhi terbentuknya detak jantung yang cepat, yang seringkali terjadi selama kegiatan fisik atau kegembiraan emosional. Ketika detak jantung terlalu cepat, otak bisa tidak menerima cukup darah. Akibatnya adalah kehilangan kesadaran. Beberapa orang dengan sindrom long QT juga terlahir tuli. Tetapi sekitar sepertiga orang tidak mengalami gejala-gejala. Sindrom long QT bisa menyebabkan kematian tiba-tiba pada usia muda.
DIAGNOSA
Dokter bisa menganjurkan electrocardiogfrafi (ECG) untuk anak atau orang dewasa muda yang mengalami hilangnya kesadaran secara tiba-tiba dan tidak dapat dijelaskan. Prosedur tersebut kemungkinan dilakukan dengan orang tersebut ketika istirahat atau setelah menerima obat-obatan secara infus atau orang tersebut kemungkinan diminta untuk berjalan di atas treadmill atau pedal sebuah sepeda latihan dalam prosedur yang disebut tes latihan stress.
PENGOBATAN
Beta-blocker efektif untuk kebanyakan anak dan orang dewasa. Beberapa orang dewasa bisa menerima manfaat dari mexiletine, sebuah obat antiaritmia. Untuk anak dan orang dewasa yang tidak bereaksi terhadap obat-obatan, sebuah pacemaker atau kombinasi pacemaker-internal defibrillator kemungkinan dicoba. Internal defibrillator bisa mengejutkan jantung, menghidupkan kembali orang tersebut, kapan saja jantung tersebut membentuk kelainan ritme yang mematikan. Kadangkala, sebagai alternatif, saraf pada leher dipotong dalam prosedur yang disebut cervicothoracic sympathectomy. Pemotongan saraf ini bisa membantu mencegah detak jantung cepat yang menyebabkan kematian tiba-tiba.
Tidak ada komentar:
Posting Komentar