





- Stenosis Katup Pulmoner
- Kelainan bawaan (Kelainan Kongenital)
- Kelainan Jantung Bawaan
- Defek Septum Ventrikel (VSD, Ventricular Septal De...
- Patent Ductus Arteriosus
- Stenosis Katup Aorta
- Koartasio Aorta
- Transposisi Arteri Besar
- Sindroma Hipoplastik Jantung Kiri
- Tetralogi Fallot
- Atresia Esofagus
- Penyakit Hirschprung
- Omfalokel
- Atresia Anus : Lubang Anus Tidak Terbentuk
100. Atresia Bilier
101. Kelainan Tulang & Otot Bawaan
102. Kelainan Tulang & Otot Bawaan
103. Defek Septum Atrium (ASD, Atrial Septal Defect)
104. Kelainan Otak Bawaan
105. Spina Bifida (Sumbing Tulang Belakang)
106. Kelainan Ginjal & Saluran Kemih Bawaan
107. Intersex States
108. Sindroma 4p
109. Sindroma Cri Du Chat : Tangisan Anak Seperti Suara...
110. Kekurangan Gizi (Malnutrisi)
111. Kelainan Metabolisme
112. Kelainan Mata Bawaan
113. Hydrocephalus
114. Kelainan Kelamin
115. Kelainan Dinding Abdominal(Omphalocele dan Gastros...
Stenosis Katup Pulmoner
Stenosis Katup Pulmoner adalah suatu penyempitan atau penyumbatan pada katup pulmoner.
Katup pulmoner adalah katup pada ventrikel kanan jantung, yang akan membuka untuk mengalirkan darah ke paru-paru.

PENYEBAB
Stenos pulmoner seringkali disebabkan oleh adanya gangguan pembentukan selama perkembangan janin yang penyebabnya tidak diketahui.
Penyempitan bisa terjadi pada katup pulmoner maupun di bawah katup pulmoner (pada arteri pulmonalis).
Kelainan ini bisa berdiri sendiri atau bersamaan dengan kelainan jantung lainnya, sifatnya bisa ringan maupun berat.
Ditemukan pada 1 diantara 8000 bayi.
GEJALA
Jika terjadi penyumbatan yang lebih berat, maka darah yang mengalir ke paru-paru sangat sedikit. Tekanan di ventrikel dan atrium kanan meningkat, sehingga mendorong darah yang kekurangan oksigen (yang berwarna biru) menembus ke dinding diantara atrium kiri dan kanan, lalu masuk ke dalam ventrikel kiri dan dipompa ke dalam aorta untuk dialirkan ke seluruh tubuh. Akibatnya bayi tampak biru (keadaan ini disebut sianosis).
Berat badan tidak bertambah dan anak gagal berkembang.
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik.
Pada pemeriksaan dengan stetoskop akan terdengar murmur (bunyi jantung abnormal yang terjadi karena darah menyembur melewati saluran yang sempit).
Pemeriksaan yang biasa dilakukan:
- Rontgen dada
- EKG
- Ekokardiogram
- Kateterisasi jantung
- USG Doppler.
PENGOBATAN
Jika penyakitnya sedang sampai berat, katup bisa dibuka dengan cara memasukkan sebuah selang plastik yang pada ujungnya terpasang balon melalui sebuah vena (pembuluh balik) di tungkai.
Jika terjadi sianosis, maka sebelum dilakukan pembedahan diberikan obat prostaglandin (misalnya alprostadil) agar duktus arteriosus tetap terbuka.
Pembedahan yang dilakukan bisa berupa membuat hubungan antara aorta dan arteri pulmonalis atau membuka katup pulmoner maupun keduanya. Pembedahan ini memungkinkan darah untuk tidak melewati katup yang menyempit dan mengalir ke dalam paru-paru agar kaya akan oksigen.
Pembedahan biasanya dilakukan pada usia pra-sekolah.
Jika terdapat kelainan bentuk katup, maka dilakukan pembedahan untuk kembali membentuk katup pulmoner.
Katup pulmoner adalah katup pada ventrikel kanan jantung, yang akan membuka untuk mengalirkan darah ke paru-paru.
PENYEBAB
Stenos pulmoner seringkali disebabkan oleh adanya gangguan pembentukan selama perkembangan janin yang penyebabnya tidak diketahui.
Penyempitan bisa terjadi pada katup pulmoner maupun di bawah katup pulmoner (pada arteri pulmonalis).
Kelainan ini bisa berdiri sendiri atau bersamaan dengan kelainan jantung lainnya, sifatnya bisa ringan maupun berat.
Ditemukan pada 1 diantara 8000 bayi.
GEJALA
Jika terjadi penyumbatan yang lebih berat, maka darah yang mengalir ke paru-paru sangat sedikit. Tekanan di ventrikel dan atrium kanan meningkat, sehingga mendorong darah yang kekurangan oksigen (yang berwarna biru) menembus ke dinding diantara atrium kiri dan kanan, lalu masuk ke dalam ventrikel kiri dan dipompa ke dalam aorta untuk dialirkan ke seluruh tubuh. Akibatnya bayi tampak biru (keadaan ini disebut sianosis).
Berat badan tidak bertambah dan anak gagal berkembang.
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik.
Pada pemeriksaan dengan stetoskop akan terdengar murmur (bunyi jantung abnormal yang terjadi karena darah menyembur melewati saluran yang sempit).
Pemeriksaan yang biasa dilakukan:
- Rontgen dada
- EKG
- Ekokardiogram
- Kateterisasi jantung
- USG Doppler.
PENGOBATAN
Jika penyakitnya sedang sampai berat, katup bisa dibuka dengan cara memasukkan sebuah selang plastik yang pada ujungnya terpasang balon melalui sebuah vena (pembuluh balik) di tungkai.
Jika terjadi sianosis, maka sebelum dilakukan pembedahan diberikan obat prostaglandin (misalnya alprostadil) agar duktus arteriosus tetap terbuka.
Pembedahan yang dilakukan bisa berupa membuat hubungan antara aorta dan arteri pulmonalis atau membuka katup pulmoner maupun keduanya. Pembedahan ini memungkinkan darah untuk tidak melewati katup yang menyempit dan mengalir ke dalam paru-paru agar kaya akan oksigen.
Pembedahan biasanya dilakukan pada usia pra-sekolah.
Jika terdapat kelainan bentuk katup, maka dilakukan pembedahan untuk kembali membentuk katup pulmoner.
Kelainan bawaan (Kelainan Kongenital)
Kelainan Bawaan (Kelainan Kongenital)
adalah suatu kelainan pada struktur, fungsi maupun metabolisme tubuh
yang ditemukan pada bayi ketika dia dilahirkan.
Sekitar 3-4% bayi baru lahir memiliki kelainan bawaan yang berat.
Beberapa kelainan baru ditemukan pada saat anak mulai tumbuh, yaitu sekitar 7,5% terdiagnosis ketika anak berusia 5 tahun, tetapi kebanyakan bersifat ringan.
PENYEBAB
Kebanyakan bayi yang lahir dengan kelainan bawaan memiliki orang tua yang jelas-jelas tidak memiliki gangguan kesehatan maupun faktor resiko. Seorang wanita hamil yang telah mengikuti semua nasihat dokternya agar kelak melahirkan bayi yang sehat, mungkin saja nanti melahirkan bayi yang memilii kelainan bawaan.
60% kasus kelainan bawaan penyebabnya tidak diketahui; sisanya disebabkan oleh faktor lingkungan atau genetik atau kombinasi dari keduanya.
Kelainan struktur atau kelainan metabolisme terjadi akibat:
- hilangnya bagian tubuh tertentu
- kelainan pembentukan bagian tubuh tertentu
- kelainan bawaan pada kimia tubuh.
Kelainan struktur utama yang paling sering ditemukan adalah kelainan jantung, diikuti oleh spina bifida dan hipospadia.
Kelainan metabolisme biasanya berupa hilangnya enzim atau tidak sempurnanya pembentukan enzim. Kelainan ini berbahaya bahkan bisa berakibat fatal, tetapi biasanya tidak menimbulkan gangguan yang nyata pada anak.
Contoh dari kelainan metabolisme adalah penyakit Tay-Sachs (penyakit fatal pada sistem saraf pusat) dan fenilketonuria.
Penyebab lain dari kelainan bawaan adalah:
# Pemakaian alkohol oleh ibu hamil
Pemakaian alkohol oleh ibu hamil bisa menyebabkan sindroma alkohol pada janin dan obat-obat tertentu yang diminum oleh ibu hamil juga bisa menyebakan kelainan bawaan.
# Penyakit Rh, terjadi jika ibu dan bayi memiliki faktor Rh yang berbeda.
Beberapa faktor yang dapat menyebabkan meningkatnya resiko kelainan bawaan:
1. Teratogenik
Teratogen adalah setiap faktor atau bahan yang bisa menyebabkan atau meningkatkan resiko suatu kelainan bawaan.
Radiasi, obat tertentu dan racun merupakan teratogen.
Secara umum, seorang wanita hamil sebaiknya:
- mengkonsultasikan dengan dokternya setiap obat yang dia minum
- berhenti merokok
- tidak mengkonsumsi alkohol
- tidak menjalani pemeriksaan rontgen kecuali jika sangat mendesak.
Infeksi pada ibu hamil juga bisa merupakan teratogen. Beberapa infeksi selama kehamilan yang dapat menyebabkan sejumlah kelainan bawaan:
- Sindroma rubella kongenital ditandai dengan gangguan penglihatan atau pendengaran, kelainan jantung, keterbelakangan mental dan cerebral palsy
- Infeksi toksoplasmosis pada ibu hamil bisa menyebabkan infeksi mata yang bisa berakibat fatal, gangguan pendengaran, ketidakmampuan belajar, pembesaran hati atau limpa, keterbelakangan mental dan cerebral palsy
- Infeksi virus herpes genitalis pada ibu hamil, jika ditularkan kepada bayinya sebelum atau selama proses persalinan berlangsung, bisa menyebabkan kerusakan otak, cerebral palsy, gangguan penglihatan atau pendengaran serta kematian bayi
- Penyakit ke-5 bisa menyebabkan sejenis anemia yang berbahaya, gagal jantung dan kematian janin
- Sindroma varicella kongenital disebabkan oleh cacar air dan bisa menyebabkan terbentuknya jaringan parut pada otot dan tulang, kelainan bentuk dan kelumpuhan pada anggota gerak, kepala yang berukuran lebih kecil dari normal, kebutaan, kejang dan keterbelakangan mental.
2. Gizi
Menjaga kesehatan janin tidak hanya dilakukan dengan menghindari teratogen, tetapi juga dengan mengkonsumsi gizi yang baik.
Salah satu zat yang penting untuk pertumbuhan janin adalah asam folat. Kekurangan asam folat bisa meningkatkan resiko terjadinya spina bifida atau kelainan tabung saraf lainnya. Karena spina bifida bisa terjadi sebelum seorang wanita menyadari bahwa dia hamil, maka setiap wanita usia subur sebaiknya mengkonsumsi asam folat minimal sebanyak 400 mikrogram/hari.
3. Faktor fisik pada rahim
Di dalam rahim, bayi terendam oleh cairan ketuban yang juga merupakan pelindung terhadap cedera.
Jumlah cairan ketuban yang abnormal bisa menyebabkan atau menunjukkan adanya kelainan bawaan.
Cairan ketuban yang terlalu sedikit bisa mempengaruhi pertumbuhan paru-paru dan anggota gerak tubuh atau bisa menunjukkan adanya kelainan ginjal yang memperlambat proses pembentukan air kemih.
Penimbunan cairan ketuban terjadi jika janin mengalami gangguan menelan, yang bisa disebabkan oleh kelainan otak yang berat (misalnya anensefalus atau atresia esofagus).
4. Faktor genetik dan kromosom
Genetik memegang peran penting dalam beberapa kelainan bawaan. Beberapa kelainan bawaan merupakan penyakit keturunan yang diwariskan melalui gen yang abnormal dari salah satu atau kedua orang tua.
Gen adalah pembawa sifat individu yang terdapat di dalam kromosom setiap sel di dalam tubuh manusia. Jika 1 gen hilang atau cacat, bisa terjadi kelainan bawaan.
Pola pewarisan kelainan genetik:
1. Autosom dominan
Jika suatu kelainan atau penyakit timbul meskipun hanya terdapat 1 gen yang cacat dari salah satu orang tuanya, maka keadaannya disebut autosom dominan.
Contohnya adalah akondroplasia dan sindroma Marfan.
2. Autosom resesif
Jika untuk terjadinya suatu kelainan bawaan diperlukan 2 gen yang masing-masing berasal dari kedua orang tua, maka keadaannya disebut autosom resesif.
Contohnya adalah penyakit Tay-Sachs atau kistik fibrosis.
3. X-linked
Jika seorang anak laki-laki mendapatkan kelainan dari gen yang berasal dari ibunya, maka keadaannya disebut X-linked, karena gen tersebut dibawa oleh kromosom X.
Laki-laki hanya memiliki 1 kromosom X yang diterima dari ibunya (perempuan memiliki 2 kromosom X, 1 berasal dari ibu dan 1 berasal dari ayah), karena itu gen cacat yang dibawa oleh kromosom X akan menimbulkan kelainan karena laki-laki tidak memiliki salinan yang normal dari gen tersebut.
Contohnya adalah hemofilia dan buta warna.
Kelainan pada jumlah ataupun susunan kromosom juga bisa menyebabkan kelainan bawaan.
Suatu kesalahan yang terjadi selama pembentukan sel telur atau sperma bisa menyebabkan bayi terlahir dengan kromosom yang terlalu banyak atau terlalu sedikit, atau bayi terlahir dengan kromosom yang telah mengalami kerusakan.
Contoh dari kelainan bawaan akibat kelainan pada kromosom adalah sindroma Down.
Semakin tua usia seorang wanita ketika hamil (terutama diatas 35 tahun) maka semakin besar kemungkinan terjadinya kelainan kromosom pada janin yang dikandungnya.
Kelainan bawaan yang lainnya disebabkan oleh mutasi genetik (perubahan pada gen yang bersifat spontan dan tidak dapat dijelaskan). Meskipun bisa dilakukan berbagai tindakan untuk mencegah terjadinya kelainan bawaan, ada satu hal yang perlu diingat yaitu bahwa suatu kelainan bawaan bisa saja terjadi meskipun tidak ditemukan riwayat kelainan bawaan baik dalam keluarga ayah ataupun ibu, atau meskipun orang tua sebelumnya telah melahirkan anak-anak yang sehat.
GEJALA
Kelainan bawaan menyebabkan gangguan fisik atau mental atau bisa berakibat fatal.
Terdapat lebih dari 4.000 jenis kelainan bawaan, mulai dari yang ringan sampai yang serius, dan meskipun banyak diantaranya yang dapat diobati maupun disembuhkan, tetapi kelainan bawaan tetap merupakan penyebab utama dari kematian pada tahun pertama kehidupan bayi.
Beberapa kelainan bawaan yang sering ditemukan:
1. Celah bibir atau langit-langit mulut (sumbing)
Terjadi jika selama masa perkembangan janin, jaringan mulut atau bibir tidak terbentuk sebagaimana mestinya.
Bibir sumbing adalah suatu celah diantara bibir bagian atas dengan hidung.
Langit-langit sumbing adalah suatu celah diantara langit-langit mulut dengan rongga hidung.
2. Defek tabung saraf
Terjadi pada awal kehamilan, yaitu pada saat terbentuknya bakal otak dan korda spinalis. Dalam keadaan normal, struktur tersebut melipat membentuk tabung pada hari ke 29 setelah pembuahan. Jika tabung tidak menutup secara sempurna, maka akan terjadi defek tabung saraf.
Bayi yang memiliki kelainan ini banyak yang meninggal di dalam kandungan atau meninggal segera setelah lahir.
2 macam defek tabung saraf yang paling sering ditemukan:
- Spina bifida, terjadi jika kolumna spinalis tidak menutup secara sempurna di sekeliling korda spinalis.
- Anensefalus, terjadi jika beberapa bagian otak tidak terbentuk.
3. Kelainan jantung
- Defek septum atrium dan ventrikel (terdapat lubang pada dinding yang meimsahkan jantung kiri dan kanan)
- Patent ductus arteriosus (terjadi jika pembuluh darah yang penting pada sirkulasi janin ketika masih berada di dalam rahim; setelah bayi lahir, tidak menutup sebagaimana mestinya)
- Stenosis katup aorta atau pulmonalis (penyempitan katup aorta atau katup pulmonalis)
- Koartasio aorta (penyempitan aorta)
- Transposisi arteri besar (kelainan letak aorta dan arteri pulmonalis)
- Sindroma hipoplasia jantung kiri (bagian jantung yang memompa darah ke seluruh tubuh tidak terbentuk sempurna)
- Tetralogi Fallot (terdiri dari stenosis katup pulmonalis, defek septum ventrikel, transposisi arteri besar dan hipertrofi ventrikel kanan).
Pemakaian obat tertentu pada kehamilan trimester pertama berperan dalam terjadinya kelainan jantung bawaan (misalnya obat anti-kejang fenitoin, talidomid dan obat kemoterapi).
Penyebab lainnya adalah pemakaian alkohol, rubella dan diabetes selama hamil.
4. Cerebral palsy
Biasanya baru diketahui beberapa minggu atau beberapa bulan setelah bayi lahir, tergantung kepada beratnya kelainan.
5. Clubfoot
Istilah clubfoot digunakan untuk menggambarkan sekumpulan kelainan struktur pada kaki dan pergelangan kaki, dimana terjadi kelainan pada pembentukan tulang, sendi, otot dan pembuluh darah.
6. Dislokasi panggul bawaan
Terjadi jika ujung tulang paha tidak terletak di dalam kantung panggul.
7. Hipotiroidisme kongenital
Terjadi jika bayi tidak memiliki kelenjar tiroid atau jika kelenjar tiroid tidak terbentuk secara sempurna.
8. Fibrosis kistik
Penyakit ini terutama menyerang sistem pernafasan dan saluran pencernaan. Tubuh tidak mampu membawa klorida dari dalam sel ke permukaan organ sehingga terbentuk lendir yang kental dan lengket.
9. Defek saluran pencernaan
Saluran pencernaan terdiri dari kerongkongan, lambung, usus halus dan usus besar, rektum serta anus.
Diantaranya adalah:
- Atresia esofagus (kerongkongan tidak terbentuk sempurna)
- Hernia diafragmatika
- Stenosis pilorus
- Penyakit Hirschsprung
- Gastroskisis dan omfalokel
- Atresia anus
- Atresia bilier
10. Sindroma Down
Merupakan sekumpulan kelainan yang terjadi pada anak-anak yang dilahirkan dengan kelebihan kromosom nomor 21 pada sel-selnya.
Mereka mengalami keterbelakangan mental dan memiliki wajah dan gambaran fisik lainnya yang khas; kelainan ini sering disertai dengan kelainan jantung.
11. Fenilketonuria
Merupakan suatu penyakit yang mempengaruhi pengolahan protein oleh tubuh dan bisa menyebabkan keterbelakangan mental.
Bayi yang terlahir dengan fenilketonuria tampak normal, tetapi jika tidak diobati mereka akan mengalami gangguan perkembangan yang baru terlihat ketika usianya mencapai 1 tahun.
12. Sindroma X yang rapuh
Sindroma ini ditandai dengan gangguan mental, mulai dari ketidakmampuan belajar sampai keterbelakangan mental, perilaku autis dan gangguan pemusatan perhatian serta hiperaktivitas.
Gambaran fisiknya khas, yaitu wajahnya panjang, telinganya lebar, kakinya datar dan persendiannya sangat lentur (terutama sendi pada jari tangan).
Sindroma ini lebih banyak ditemukan pada anak laki-laki.
13. Distrofi otot
Distrofi otot adalah suatu istilah yang digunakan untuk menggambarkan lebih dari 40 macam penyakit otot yang berlainan, yang kesemuanya ditandai dengan kelemahan dan kemunduran yang progresif dari otot-otot yang mengendalikan pergerakan.
14. Anemia sel sabit
Merupakan suatu kelainan sel darah merah yang memiliki bentuk abnormal (seperti bulan sabit), yang menyebabkan anemia kronis, serangan nyeri dan gangguan kesehatan lainnya.
15. Penyakit Tay-Sachs
Penyakit ini menyerang sistem saraf pusat dan menyebabkan kebutaan, demensia, kelumpuhan, kejang dan ketulian.
16. Sindroma alkohol pada janin
Sindroma in ditandai dengan keterlambatan pertumbuhan, keterbelakangan mental, kelainan pada wajah dan kelainan pada sistem saraf pusat.
DIAGNOSA
Selama menjalani perawatan prenatal, ada beberapa jenis tes yang ditawarkan kepada semua wanita hamil (tes skrining) dan ada pula beberapa jenis tes yang ditawarkan hanya kepada wanita/pasangan suami-istri yang memiliki faktor resiko (tes diagnostik).
Tidak ada tes yang sempurna. Seorang bayi mungkin saja terlahir dengan kelainan bawaan meskipun hasil tesnya negatif. Jika tes memberikan hasil yang positif, biasanya perlu dilakukan tes lebih lanjut.
Tes skrining
Tes skrining dilakukan meskipun seorang wanita hamil tidak memiliki gejala maupun faktor resiko.
Bila tes skrining menunjukkan hasil positif, dianjurkan untuk menjalani tes diagnostik.
Skrining prenatal bisa membantu menentukan adanya infeksi atau keadaan lain pada ibu yang berbahaya bagi janin dan membantu menentukan adanya kelainan bawaan tertentu pada janin.
Tes skrining terdiri dari:
# Pemeriksaan darah
# Pemeriksaan USG.
Tes diagnostik
Tes diagnostik biasanya dilakukan jika tes skrining memberikan hasil positif atau jika wanita hamil memiliki faktor resiko.
Tes diagnostik terdiri dari:
# Amniosentesis
# Contoh vili korion
# Contoh darah janin
# Pemeriksaan USG yang lebih mendetil.
Kelainan bawaan yang bisa diketahui melalui skrining prenatal adalah:
# Defek tabung saraf (spina bifida, anensefalus)
# Sindroma Down
# Kelainan kromosom lainnya
# Kelainan metabolisme yang diturunkan
# Kelainan jantung bawaan
# Kelainan bentuk saluran pencernaan dan ginjal
# Sumbing bibir atau langit-langit mulut
# Kelainan bawaan tertentu pada anggota gerak
# Tumor bawaan.
PENCEGAHAN
Beberapa kelainan bawaan tidak dapat dicegah, tetapi ada beberapa hal yang dapat dilakukan untuk mengurangi resiko terjadinya kelainan bawaan:
# Tidak merokok dan menghindari asap rokok
# Menghindari alkohol
# Menghindari obat terlarang
# Memakan makanan yang bergizi dan mengkonsumsi vitamin prenatal
# Melakukan olah raga dan istirahat yang cukup
# Melakukan pemeriksaan prenatal secara rutin
# Mengkonsumsi suplemen asam folat
# Menjalani vaksinasi sebagai perlindungan terhadap infeksi
# Menghindari zat-zat yang berbahaya.
Vaksinasi
Vaksinasi membantu mencegah penyakit akibat infeksi. Meskipun semua vaksin aman diberikan pada masa hamil, tetapi akan lebih baik jika semua vaksin yang dibutuhkan telah dilaksanakan sebelum hamil.
Seorang wanita sebaiknya menjalani vaksinasi berikut:
1. Minimal 3 bulan sebelum hamil : MMR
2. Minimal 1 bulan sebelum hamil : varicella
3. Aman diberikan pada saat hamil
- Booster tetanus-difteri (setiap 10 tahun)
- Vaksin hepatitis A
- Vaksin hepatitis B
- Vaksin influenza (jika pada musim flu kehamilan akan memasuki trimester kedua atau ketiga)
- Vaksin pneumokokus.
Zat yang berbahaya
Beberapa zat yang berbahaya selama kehamilan:
# Alkohol
# Androgen dan turunan testosteron (misalnya danazol)
# Angiotensin-converting enzyme (ACE) inhibitors (misalnya enalapril, captopril)
# Turunan kumarin (misalnya warfarin)
# Carbamazepine
# Antagonis asam folat (misalnya metotrexat dan aminopterin)
# Cocain
# Dietilstilbestrol
# Timah hitam
# Lithium
# Merkuri organik
# Phenitoin
# Streptomycin dan kanamycin
# Tetrasyclin
# Talidomide
# Trimethadion dan paramethadion
# Asam valproat
# Vitamin A dan turunannya (misalnya isotretinoin, etretinat dan retinoid)
# Infeksi
# Radiasi.
Meskipun bisa dilakukan berbagai tindakan untuk mencegah terjadinya kelainan bawaan, ada satu hal yang perlu diingat yaitu bahwa suatu kelainan bawaan bisa saja terjadi meskipun tidak ditemukan riwayat kelainan bawaan baik dalam keluarga ayah ataupun ibu, atau meskipun orang tua sebelumnya telah melahirkan anak-anak yang sehat.
Sekitar 3-4% bayi baru lahir memiliki kelainan bawaan yang berat.
Beberapa kelainan baru ditemukan pada saat anak mulai tumbuh, yaitu sekitar 7,5% terdiagnosis ketika anak berusia 5 tahun, tetapi kebanyakan bersifat ringan.
PENYEBAB
Kebanyakan bayi yang lahir dengan kelainan bawaan memiliki orang tua yang jelas-jelas tidak memiliki gangguan kesehatan maupun faktor resiko. Seorang wanita hamil yang telah mengikuti semua nasihat dokternya agar kelak melahirkan bayi yang sehat, mungkin saja nanti melahirkan bayi yang memilii kelainan bawaan.
60% kasus kelainan bawaan penyebabnya tidak diketahui; sisanya disebabkan oleh faktor lingkungan atau genetik atau kombinasi dari keduanya.
Kelainan struktur atau kelainan metabolisme terjadi akibat:
- hilangnya bagian tubuh tertentu
- kelainan pembentukan bagian tubuh tertentu
- kelainan bawaan pada kimia tubuh.
Kelainan struktur utama yang paling sering ditemukan adalah kelainan jantung, diikuti oleh spina bifida dan hipospadia.
Kelainan metabolisme biasanya berupa hilangnya enzim atau tidak sempurnanya pembentukan enzim. Kelainan ini berbahaya bahkan bisa berakibat fatal, tetapi biasanya tidak menimbulkan gangguan yang nyata pada anak.
Contoh dari kelainan metabolisme adalah penyakit Tay-Sachs (penyakit fatal pada sistem saraf pusat) dan fenilketonuria.
Penyebab lain dari kelainan bawaan adalah:
# Pemakaian alkohol oleh ibu hamil
Pemakaian alkohol oleh ibu hamil bisa menyebabkan sindroma alkohol pada janin dan obat-obat tertentu yang diminum oleh ibu hamil juga bisa menyebakan kelainan bawaan.
# Penyakit Rh, terjadi jika ibu dan bayi memiliki faktor Rh yang berbeda.
Beberapa faktor yang dapat menyebabkan meningkatnya resiko kelainan bawaan:
1. Teratogenik
Teratogen adalah setiap faktor atau bahan yang bisa menyebabkan atau meningkatkan resiko suatu kelainan bawaan.
Radiasi, obat tertentu dan racun merupakan teratogen.
Secara umum, seorang wanita hamil sebaiknya:
- mengkonsultasikan dengan dokternya setiap obat yang dia minum
- berhenti merokok
- tidak mengkonsumsi alkohol
- tidak menjalani pemeriksaan rontgen kecuali jika sangat mendesak.
Infeksi pada ibu hamil juga bisa merupakan teratogen. Beberapa infeksi selama kehamilan yang dapat menyebabkan sejumlah kelainan bawaan:
- Sindroma rubella kongenital ditandai dengan gangguan penglihatan atau pendengaran, kelainan jantung, keterbelakangan mental dan cerebral palsy
- Infeksi toksoplasmosis pada ibu hamil bisa menyebabkan infeksi mata yang bisa berakibat fatal, gangguan pendengaran, ketidakmampuan belajar, pembesaran hati atau limpa, keterbelakangan mental dan cerebral palsy
- Infeksi virus herpes genitalis pada ibu hamil, jika ditularkan kepada bayinya sebelum atau selama proses persalinan berlangsung, bisa menyebabkan kerusakan otak, cerebral palsy, gangguan penglihatan atau pendengaran serta kematian bayi
- Penyakit ke-5 bisa menyebabkan sejenis anemia yang berbahaya, gagal jantung dan kematian janin
- Sindroma varicella kongenital disebabkan oleh cacar air dan bisa menyebabkan terbentuknya jaringan parut pada otot dan tulang, kelainan bentuk dan kelumpuhan pada anggota gerak, kepala yang berukuran lebih kecil dari normal, kebutaan, kejang dan keterbelakangan mental.
2. Gizi
Menjaga kesehatan janin tidak hanya dilakukan dengan menghindari teratogen, tetapi juga dengan mengkonsumsi gizi yang baik.
Salah satu zat yang penting untuk pertumbuhan janin adalah asam folat. Kekurangan asam folat bisa meningkatkan resiko terjadinya spina bifida atau kelainan tabung saraf lainnya. Karena spina bifida bisa terjadi sebelum seorang wanita menyadari bahwa dia hamil, maka setiap wanita usia subur sebaiknya mengkonsumsi asam folat minimal sebanyak 400 mikrogram/hari.
3. Faktor fisik pada rahim
Di dalam rahim, bayi terendam oleh cairan ketuban yang juga merupakan pelindung terhadap cedera.
Jumlah cairan ketuban yang abnormal bisa menyebabkan atau menunjukkan adanya kelainan bawaan.
Cairan ketuban yang terlalu sedikit bisa mempengaruhi pertumbuhan paru-paru dan anggota gerak tubuh atau bisa menunjukkan adanya kelainan ginjal yang memperlambat proses pembentukan air kemih.
Penimbunan cairan ketuban terjadi jika janin mengalami gangguan menelan, yang bisa disebabkan oleh kelainan otak yang berat (misalnya anensefalus atau atresia esofagus).
4. Faktor genetik dan kromosom
Genetik memegang peran penting dalam beberapa kelainan bawaan. Beberapa kelainan bawaan merupakan penyakit keturunan yang diwariskan melalui gen yang abnormal dari salah satu atau kedua orang tua.
Gen adalah pembawa sifat individu yang terdapat di dalam kromosom setiap sel di dalam tubuh manusia. Jika 1 gen hilang atau cacat, bisa terjadi kelainan bawaan.
Pola pewarisan kelainan genetik:
1. Autosom dominan
Jika suatu kelainan atau penyakit timbul meskipun hanya terdapat 1 gen yang cacat dari salah satu orang tuanya, maka keadaannya disebut autosom dominan.
Contohnya adalah akondroplasia dan sindroma Marfan.
2. Autosom resesif
Jika untuk terjadinya suatu kelainan bawaan diperlukan 2 gen yang masing-masing berasal dari kedua orang tua, maka keadaannya disebut autosom resesif.
Contohnya adalah penyakit Tay-Sachs atau kistik fibrosis.
3. X-linked
Jika seorang anak laki-laki mendapatkan kelainan dari gen yang berasal dari ibunya, maka keadaannya disebut X-linked, karena gen tersebut dibawa oleh kromosom X.
Laki-laki hanya memiliki 1 kromosom X yang diterima dari ibunya (perempuan memiliki 2 kromosom X, 1 berasal dari ibu dan 1 berasal dari ayah), karena itu gen cacat yang dibawa oleh kromosom X akan menimbulkan kelainan karena laki-laki tidak memiliki salinan yang normal dari gen tersebut.
Contohnya adalah hemofilia dan buta warna.
Kelainan pada jumlah ataupun susunan kromosom juga bisa menyebabkan kelainan bawaan.
Suatu kesalahan yang terjadi selama pembentukan sel telur atau sperma bisa menyebabkan bayi terlahir dengan kromosom yang terlalu banyak atau terlalu sedikit, atau bayi terlahir dengan kromosom yang telah mengalami kerusakan.
Contoh dari kelainan bawaan akibat kelainan pada kromosom adalah sindroma Down.
Semakin tua usia seorang wanita ketika hamil (terutama diatas 35 tahun) maka semakin besar kemungkinan terjadinya kelainan kromosom pada janin yang dikandungnya.
Kelainan bawaan yang lainnya disebabkan oleh mutasi genetik (perubahan pada gen yang bersifat spontan dan tidak dapat dijelaskan). Meskipun bisa dilakukan berbagai tindakan untuk mencegah terjadinya kelainan bawaan, ada satu hal yang perlu diingat yaitu bahwa suatu kelainan bawaan bisa saja terjadi meskipun tidak ditemukan riwayat kelainan bawaan baik dalam keluarga ayah ataupun ibu, atau meskipun orang tua sebelumnya telah melahirkan anak-anak yang sehat.
GEJALA
Kelainan bawaan menyebabkan gangguan fisik atau mental atau bisa berakibat fatal.
Terdapat lebih dari 4.000 jenis kelainan bawaan, mulai dari yang ringan sampai yang serius, dan meskipun banyak diantaranya yang dapat diobati maupun disembuhkan, tetapi kelainan bawaan tetap merupakan penyebab utama dari kematian pada tahun pertama kehidupan bayi.
Beberapa kelainan bawaan yang sering ditemukan:
1. Celah bibir atau langit-langit mulut (sumbing)
Terjadi jika selama masa perkembangan janin, jaringan mulut atau bibir tidak terbentuk sebagaimana mestinya.
Bibir sumbing adalah suatu celah diantara bibir bagian atas dengan hidung.
Langit-langit sumbing adalah suatu celah diantara langit-langit mulut dengan rongga hidung.
2. Defek tabung saraf
Terjadi pada awal kehamilan, yaitu pada saat terbentuknya bakal otak dan korda spinalis. Dalam keadaan normal, struktur tersebut melipat membentuk tabung pada hari ke 29 setelah pembuahan. Jika tabung tidak menutup secara sempurna, maka akan terjadi defek tabung saraf.
Bayi yang memiliki kelainan ini banyak yang meninggal di dalam kandungan atau meninggal segera setelah lahir.
2 macam defek tabung saraf yang paling sering ditemukan:
- Spina bifida, terjadi jika kolumna spinalis tidak menutup secara sempurna di sekeliling korda spinalis.
- Anensefalus, terjadi jika beberapa bagian otak tidak terbentuk.
3. Kelainan jantung
- Defek septum atrium dan ventrikel (terdapat lubang pada dinding yang meimsahkan jantung kiri dan kanan)
- Patent ductus arteriosus (terjadi jika pembuluh darah yang penting pada sirkulasi janin ketika masih berada di dalam rahim; setelah bayi lahir, tidak menutup sebagaimana mestinya)
- Stenosis katup aorta atau pulmonalis (penyempitan katup aorta atau katup pulmonalis)
- Koartasio aorta (penyempitan aorta)
- Transposisi arteri besar (kelainan letak aorta dan arteri pulmonalis)
- Sindroma hipoplasia jantung kiri (bagian jantung yang memompa darah ke seluruh tubuh tidak terbentuk sempurna)
- Tetralogi Fallot (terdiri dari stenosis katup pulmonalis, defek septum ventrikel, transposisi arteri besar dan hipertrofi ventrikel kanan).
Pemakaian obat tertentu pada kehamilan trimester pertama berperan dalam terjadinya kelainan jantung bawaan (misalnya obat anti-kejang fenitoin, talidomid dan obat kemoterapi).
Penyebab lainnya adalah pemakaian alkohol, rubella dan diabetes selama hamil.
4. Cerebral palsy
Biasanya baru diketahui beberapa minggu atau beberapa bulan setelah bayi lahir, tergantung kepada beratnya kelainan.
5. Clubfoot
Istilah clubfoot digunakan untuk menggambarkan sekumpulan kelainan struktur pada kaki dan pergelangan kaki, dimana terjadi kelainan pada pembentukan tulang, sendi, otot dan pembuluh darah.
6. Dislokasi panggul bawaan
Terjadi jika ujung tulang paha tidak terletak di dalam kantung panggul.
7. Hipotiroidisme kongenital
Terjadi jika bayi tidak memiliki kelenjar tiroid atau jika kelenjar tiroid tidak terbentuk secara sempurna.
8. Fibrosis kistik
Penyakit ini terutama menyerang sistem pernafasan dan saluran pencernaan. Tubuh tidak mampu membawa klorida dari dalam sel ke permukaan organ sehingga terbentuk lendir yang kental dan lengket.
9. Defek saluran pencernaan
Saluran pencernaan terdiri dari kerongkongan, lambung, usus halus dan usus besar, rektum serta anus.
Diantaranya adalah:
- Atresia esofagus (kerongkongan tidak terbentuk sempurna)
- Hernia diafragmatika
- Stenosis pilorus
- Penyakit Hirschsprung
- Gastroskisis dan omfalokel
- Atresia anus
- Atresia bilier
10. Sindroma Down
Merupakan sekumpulan kelainan yang terjadi pada anak-anak yang dilahirkan dengan kelebihan kromosom nomor 21 pada sel-selnya.
Mereka mengalami keterbelakangan mental dan memiliki wajah dan gambaran fisik lainnya yang khas; kelainan ini sering disertai dengan kelainan jantung.
11. Fenilketonuria
Merupakan suatu penyakit yang mempengaruhi pengolahan protein oleh tubuh dan bisa menyebabkan keterbelakangan mental.
Bayi yang terlahir dengan fenilketonuria tampak normal, tetapi jika tidak diobati mereka akan mengalami gangguan perkembangan yang baru terlihat ketika usianya mencapai 1 tahun.
12. Sindroma X yang rapuh
Sindroma ini ditandai dengan gangguan mental, mulai dari ketidakmampuan belajar sampai keterbelakangan mental, perilaku autis dan gangguan pemusatan perhatian serta hiperaktivitas.
Gambaran fisiknya khas, yaitu wajahnya panjang, telinganya lebar, kakinya datar dan persendiannya sangat lentur (terutama sendi pada jari tangan).
Sindroma ini lebih banyak ditemukan pada anak laki-laki.
13. Distrofi otot
Distrofi otot adalah suatu istilah yang digunakan untuk menggambarkan lebih dari 40 macam penyakit otot yang berlainan, yang kesemuanya ditandai dengan kelemahan dan kemunduran yang progresif dari otot-otot yang mengendalikan pergerakan.
14. Anemia sel sabit
Merupakan suatu kelainan sel darah merah yang memiliki bentuk abnormal (seperti bulan sabit), yang menyebabkan anemia kronis, serangan nyeri dan gangguan kesehatan lainnya.
15. Penyakit Tay-Sachs
Penyakit ini menyerang sistem saraf pusat dan menyebabkan kebutaan, demensia, kelumpuhan, kejang dan ketulian.
16. Sindroma alkohol pada janin
Sindroma in ditandai dengan keterlambatan pertumbuhan, keterbelakangan mental, kelainan pada wajah dan kelainan pada sistem saraf pusat.
DIAGNOSA
Selama menjalani perawatan prenatal, ada beberapa jenis tes yang ditawarkan kepada semua wanita hamil (tes skrining) dan ada pula beberapa jenis tes yang ditawarkan hanya kepada wanita/pasangan suami-istri yang memiliki faktor resiko (tes diagnostik).
Tidak ada tes yang sempurna. Seorang bayi mungkin saja terlahir dengan kelainan bawaan meskipun hasil tesnya negatif. Jika tes memberikan hasil yang positif, biasanya perlu dilakukan tes lebih lanjut.
Tes skrining
Tes skrining dilakukan meskipun seorang wanita hamil tidak memiliki gejala maupun faktor resiko.
Bila tes skrining menunjukkan hasil positif, dianjurkan untuk menjalani tes diagnostik.
Skrining prenatal bisa membantu menentukan adanya infeksi atau keadaan lain pada ibu yang berbahaya bagi janin dan membantu menentukan adanya kelainan bawaan tertentu pada janin.
Tes skrining terdiri dari:
# Pemeriksaan darah
# Pemeriksaan USG.
Tes diagnostik
Tes diagnostik biasanya dilakukan jika tes skrining memberikan hasil positif atau jika wanita hamil memiliki faktor resiko.
Tes diagnostik terdiri dari:
# Amniosentesis
# Contoh vili korion
# Contoh darah janin
# Pemeriksaan USG yang lebih mendetil.
Kelainan bawaan yang bisa diketahui melalui skrining prenatal adalah:
# Defek tabung saraf (spina bifida, anensefalus)
# Sindroma Down
# Kelainan kromosom lainnya
# Kelainan metabolisme yang diturunkan
# Kelainan jantung bawaan
# Kelainan bentuk saluran pencernaan dan ginjal
# Sumbing bibir atau langit-langit mulut
# Kelainan bawaan tertentu pada anggota gerak
# Tumor bawaan.
PENCEGAHAN
Beberapa kelainan bawaan tidak dapat dicegah, tetapi ada beberapa hal yang dapat dilakukan untuk mengurangi resiko terjadinya kelainan bawaan:
# Tidak merokok dan menghindari asap rokok
# Menghindari alkohol
# Menghindari obat terlarang
# Memakan makanan yang bergizi dan mengkonsumsi vitamin prenatal
# Melakukan olah raga dan istirahat yang cukup
# Melakukan pemeriksaan prenatal secara rutin
# Mengkonsumsi suplemen asam folat
# Menjalani vaksinasi sebagai perlindungan terhadap infeksi
# Menghindari zat-zat yang berbahaya.
Vaksinasi
Vaksinasi membantu mencegah penyakit akibat infeksi. Meskipun semua vaksin aman diberikan pada masa hamil, tetapi akan lebih baik jika semua vaksin yang dibutuhkan telah dilaksanakan sebelum hamil.
Seorang wanita sebaiknya menjalani vaksinasi berikut:
1. Minimal 3 bulan sebelum hamil : MMR
2. Minimal 1 bulan sebelum hamil : varicella
3. Aman diberikan pada saat hamil
- Booster tetanus-difteri (setiap 10 tahun)
- Vaksin hepatitis A
- Vaksin hepatitis B
- Vaksin influenza (jika pada musim flu kehamilan akan memasuki trimester kedua atau ketiga)
- Vaksin pneumokokus.
Zat yang berbahaya
Beberapa zat yang berbahaya selama kehamilan:
# Alkohol
# Androgen dan turunan testosteron (misalnya danazol)
# Angiotensin-converting enzyme (ACE) inhibitors (misalnya enalapril, captopril)
# Turunan kumarin (misalnya warfarin)
# Carbamazepine
# Antagonis asam folat (misalnya metotrexat dan aminopterin)
# Cocain
# Dietilstilbestrol
# Timah hitam
# Lithium
# Merkuri organik
# Phenitoin
# Streptomycin dan kanamycin
# Tetrasyclin
# Talidomide
# Trimethadion dan paramethadion
# Asam valproat
# Vitamin A dan turunannya (misalnya isotretinoin, etretinat dan retinoid)
# Infeksi
# Radiasi.
Meskipun bisa dilakukan berbagai tindakan untuk mencegah terjadinya kelainan bawaan, ada satu hal yang perlu diingat yaitu bahwa suatu kelainan bawaan bisa saja terjadi meskipun tidak ditemukan riwayat kelainan bawaan baik dalam keluarga ayah ataupun ibu, atau meskipun orang tua sebelumnya telah melahirkan anak-anak yang sehat.
Kelainan Jantung Bawaan
1 dari 120 bayi terlahir dengan kelainan jantung, tetapi banyak yang sifatnya tidak berat.
Kelainan jantung bisa berupa kelainan dalam pembentukan dinding maupun katup jantung atau kelainan pada pembuluh darah yang menuju dan meninggalkan jantung.
Kelainan jantung biasanya menyebabkan darah mengalir dalam arah yang salah/abnormal, kadang tidak melewati paru-paru (tempat dimana darah diperkaya dengan oksigen). Padahal untuk pertumbuhan, perkembangan dan aktivitas yang normal diperlukan darah yang kaya akan oksigen.
Beberapa kelainan jantung menyebabkan masalah yang serius, sehingga perlu dilakukan tindakan pembedahan darurat.
Aliran darah yang abnormal biasanya menyebabkan terdengarnya murmur pada pemeriksaan dengan menggunakan stetoskop. Pemeriksaan lainnya yang biasa dilakukan untuk membantu mendiagnosis suatu kelainan jantung bawaan adalah elektrokardiografi (EKG), rontgen dada dan USG.
Kelainan jantung bisa diperbaiki melalui pembedahan, yang waktu pelaksanaannya tergantung kepada gejala dan beratnya kelainan.
Beberapa kelainan jantung yang sering ditemukan:
# Defek septum atrium dan ventrikel
# PDA (Patent Ductus Arteriosus)
# Stenosis katup aorta
# Stenosis katup pulmoner
# Koartasio aorta
# Transposisi arteri besar
# Sindroma ventrikel kiri yang tidak berkembang
# Tetralogi Fallot.
Kelainan jantung bisa berupa kelainan dalam pembentukan dinding maupun katup jantung atau kelainan pada pembuluh darah yang menuju dan meninggalkan jantung.
Kelainan jantung biasanya menyebabkan darah mengalir dalam arah yang salah/abnormal, kadang tidak melewati paru-paru (tempat dimana darah diperkaya dengan oksigen). Padahal untuk pertumbuhan, perkembangan dan aktivitas yang normal diperlukan darah yang kaya akan oksigen.
Beberapa kelainan jantung menyebabkan masalah yang serius, sehingga perlu dilakukan tindakan pembedahan darurat.
Aliran darah yang abnormal biasanya menyebabkan terdengarnya murmur pada pemeriksaan dengan menggunakan stetoskop. Pemeriksaan lainnya yang biasa dilakukan untuk membantu mendiagnosis suatu kelainan jantung bawaan adalah elektrokardiografi (EKG), rontgen dada dan USG.
Kelainan jantung bisa diperbaiki melalui pembedahan, yang waktu pelaksanaannya tergantung kepada gejala dan beratnya kelainan.
Beberapa kelainan jantung yang sering ditemukan:
# Defek septum atrium dan ventrikel
# PDA (Patent Ductus Arteriosus)
# Stenosis katup aorta
# Stenosis katup pulmoner
# Koartasio aorta
# Transposisi arteri besar
# Sindroma ventrikel kiri yang tidak berkembang
# Tetralogi Fallot.
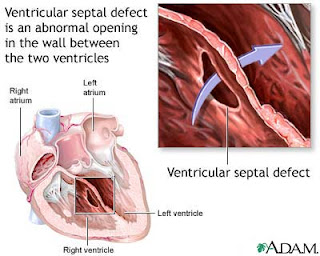
Defek Septum Ventrikel (VSD, Ventricular Septal Defect)
Defek Septum Ventrikel (VSD, Ventricular Septal Defect) adalah suatu lubang pada septum ventrikel.
Septum ventrikel adalah dinding yang memisahkan jantung bagian bawah (memisahkan ventrikel kiri dan ventrikel kanan).
PENYEBAB
Penyebabnya tidak diketahui.
VSD lebih sering ditemukan pada anak-anak dan seringkali merupakan suatu kelainan jantung bawaan.
Pada anak-anak, lubangnya sangat kecil, tidak menimbulkan gejala dan seringkali menutup dengan sendirinya sebelum anak berumur 18 tahun.
Pada kasus yang lebih berat, bisa terjadi kelainan fungsi ventrikel dan gagal jantung.
VSD bisa ditemukan bersamaan dengan kelainan jantung lainnya.
Faktor prenatal yang mungkin berhubungan dengan VSD:
- Rubella atau infeksi virus lainnya pada ibu hamil
- Gizi ibu hamil yang buruk
- Ibu yang alkoholik
- Usia ibu diatas 40 tahun
- Ibu menderita diabetes.
GEJALA
Pada kedua kelainan ini, darah dari paru-paru yang masuk ke jantung, kembali dialirkan ke paru-paru. Akibatnya jumlah darah di dalam pembuluh darah paru-paru meningkat dan menyebabkan:
- sesak nafas
- bayi mengalami kesulitan ketika menyusu
- keringat yang berlebihan
- berat badan tidak bertambah.
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik.
Dengan menggunakan stetoskop, akan terdengar murmur (bunyi jantung abnormal) yang nyaring.
Pemeriksaan yang biasa dilakukan:
# Rontgen dada
# EKG
# Ekokardiogram
# Kateterisasi jantung
# Angiografi jantung.
PENGOBATAN
Terhadap lubang yang kecil tidak perlu dilakukan penutupan, karena lubang ini seringkali menutup dengan sendirinya pada masa kanak-kanak atau remaja.
Tetapi jika lubangnya besar, meskipun gejalanya minimal, dilakukan penutupan lubang untuk mencegah terjadinya kelainan yang lebih berat. Biasanya lubang ini ditutup dengan sebuah tambalan, pada beberapa kasus hanya perlu dilakukan penjahitan tanpa harus menambal lubang.
Pembedahan biasanya dilakukan pada usia pra-sekolah (2-5 tahun)
Jika terjadi gagal jantung kongestif, diberikan obat digitalis dan diuretik.
PENCEGAHAN
Setiap wanita y Kesehatan Anak
Septum ventrikel adalah dinding yang memisahkan jantung bagian bawah (memisahkan ventrikel kiri dan ventrikel kanan).
PENYEBAB
Penyebabnya tidak diketahui.
VSD lebih sering ditemukan pada anak-anak dan seringkali merupakan suatu kelainan jantung bawaan.
Pada anak-anak, lubangnya sangat kecil, tidak menimbulkan gejala dan seringkali menutup dengan sendirinya sebelum anak berumur 18 tahun.
Pada kasus yang lebih berat, bisa terjadi kelainan fungsi ventrikel dan gagal jantung.
VSD bisa ditemukan bersamaan dengan kelainan jantung lainnya.
Faktor prenatal yang mungkin berhubungan dengan VSD:
- Rubella atau infeksi virus lainnya pada ibu hamil
- Gizi ibu hamil yang buruk
- Ibu yang alkoholik
- Usia ibu diatas 40 tahun
- Ibu menderita diabetes.
GEJALA
Pada kedua kelainan ini, darah dari paru-paru yang masuk ke jantung, kembali dialirkan ke paru-paru. Akibatnya jumlah darah di dalam pembuluh darah paru-paru meningkat dan menyebabkan:
- sesak nafas
- bayi mengalami kesulitan ketika menyusu
- keringat yang berlebihan
- berat badan tidak bertambah.
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik.
Dengan menggunakan stetoskop, akan terdengar murmur (bunyi jantung abnormal) yang nyaring.
Pemeriksaan yang biasa dilakukan:
# Rontgen dada
# EKG
# Ekokardiogram
# Kateterisasi jantung
# Angiografi jantung.
PENGOBATAN
Terhadap lubang yang kecil tidak perlu dilakukan penutupan, karena lubang ini seringkali menutup dengan sendirinya pada masa kanak-kanak atau remaja.
Tetapi jika lubangnya besar, meskipun gejalanya minimal, dilakukan penutupan lubang untuk mencegah terjadinya kelainan yang lebih berat. Biasanya lubang ini ditutup dengan sebuah tambalan, pada beberapa kasus hanya perlu dilakukan penjahitan tanpa harus menambal lubang.
Pembedahan biasanya dilakukan pada usia pra-sekolah (2-5 tahun)
Jika terjadi gagal jantung kongestif, diberikan obat digitalis dan diuretik.
PENCEGAHAN
Setiap wanita y Kesehatan Anak
Patent Ductus Arteriosus
Patent Ductus Arteriosus (PDA) adalah duktus arteriosus yang tetap terbuka.
Duktus arteriosus adalah suatu pembuluh darah yang menghubungkan aorta (pembuluh arteri besar yang mengangkut darah ke seluruh tubuh) dengan arteri pulmonalis (arteri yang membawa darah ke paru-paru), yang merupakan bagian dari peredaran darah yang normal pada janin.
Duktus arteriosus memungkinkan darah untuk tidak melewati paru-paru. Pada janin, fungsi ini penting karena janin tidak menghirup udara sehingga darah janin tidak perlu beredar melewati paru-paru agar mengandung banyak oksigen. Janin menerima oksigen dan zat makanan dari plasenta (ari-ari).
Tetapi pada saat lahir, ketika bayi mulai bernafas, duktus arteriosus akan menutup karena darah harus mengalir ke paru-paru agar mengandung banyak oksigen. Pada 95% bayi baru lahir, penutupan duktus terjadi dalam waktu 48-72 jam.
Kelainan ini bisa terjadi baik pada bayi prematur maupun pada bayi cukup umur, dan ditemukan pada 1 diantara 2500-5000 bayi.
Biasanya gejalanya ringan, tetapi akan semakin berat jika tidak diobati/diperbaiki pada usia 2 tahun.

PENYEBAB
Duktus arteriosus adalah suatu pembuluh darah yang dilapisi oleh otot dan memiliki fungsi khusus. Jika kadar oksigen di dalam darah meningkat (biasanya terjadi segera setelah bayi lahir), otot ini akan mengkerut sehingga duktus menutup.
Pada saat duktus menutup, darah dari jantung bagian kanan hanya mengalir ke paru-paru (seperti yang terjadi pada orang dewasa).
Pada beberapa anak, duktus tidak menutup atau hanya menutup sebagian. Hal ini terjadi karena tidak adanya sensor oksigen yang normal pada otot duktus atau karena kelemahan pada otot duktus.
Adapun faktor resiko terjadinya PDA adalah prematuritas dan sindroma gawat pernafasan.
GEJALA
Jika duktus tetap terbuka, darah yang seharusnya mengalir ke seluruh tubuh akan kembali ke paru-paru sehingga memenuhi pembuluh paru-paru.
Jumlah darah tambahan yang sampai ke paru-paru tergantung kepada ukuran PDA. Jika PDA sangat kecil, maka darah yang melewati PDA hanya sedikit. Pada keadaan ini, anak tidak memiliki gejala sama sekali dan tampak baik-baik saja.
PDA yang kecil dapat diketahui jika pada pemeriksaan dengan stetoskop terdengar murmur (suatu bunyi jantung ekstra yang terderngar jika darah menyembur melalui lubang yang sempit). Semakin kecil lubangnya, maka semakin sedikit darah yang mengalir dan semakin halus bunyi murmur yang terdengar.
Jika PDA memiliki lubang yang besar, maka darah dalam jumlah yang besar akan membanjiri paru-paru. Anak tampak sakit, dengan gejala berupa:
- tidak mau menyusu
- berat badannya tidak bertambah
- berkeringat
- kesulitan dalam bernafas
- denyut jantung yang cepat.
Timbulnya gejala tersebut menunjukkan telah terjadinya gagal jantung kongestif, yang seringkali terjadi pada bayi prematur.
Anak dengan PDA yang kecil tidak memiliki resiko menderita gagal jantung kongestif, tetapi tetap memiliki resiko terjadinya endokarditis.
Endokarditis adalah infeksi pada jantung, katup jantung maupun pembuluh darah jantung. Infeksi ini bisa berakibat fatal dan dapat menyebabkan kematian, stroke serta kelainan fungsi jantung.
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik.
Pada pemeriksaan dengan stetoskop seringkali terdengar murmur.
Pemeriksaan yang biasa dilakukan:
# Rontgen dada
# EKG
# Ekokardiogram.
PENGOBATAN
Jika pada saat bayi berusia beberapa minggu terjadi gagal jantung, maka segera dilakukan pembedahan.
Jika gejalanya hanya berupa murmur, maka pembedahan biasanya dilakukan pada saat anak berusia 1 tahun.
Jika tidak ada gejala, pembedahan ditunda sampai anak berumur 6 bulan-3 tahun.
Terdapat beberapa cara untuk mengatasi PDA, yang pemilihannya tergantung kepada berbagai faktor dan yang terpenting adalah usia anak:
# Bayi prematur
- Pemberian indometasin, yang secara tidak langsung akan merangsang kontraksi (pengkerutan) otot duktus.
Efek samping dari indometasin adalah perdarahan internal dan kelainan fungsi ginjal.
Jika pemberian pertama tidak berhasil dan tidak terjadi efek samping, pemberian indometasin bisa diulang.
- Pembedahan
Jika indometasin tidak efektif atau tidak dapat diberikan karena ada masalah medis lainnya, maka dilakukan pembedahan. Kedua ujung PDA diikat atau dijahit, lalu duktus dipotong.
# Bayi dan anak-anak
Pada bayi cukup umur atau anak-anak yang lebih besar, jarang terjadi gagal jantung kongestif.
Pada bayi berusia dibawah 6 bulan yang menunjukkan tanda-tanda gagal jantung, biasanya dilakukan pembedahan.
Pada anak yang berumur lebih dari 6 bulan dan tidak menunjukkan gejala, dilakukan teknik penutupan transkateter.
Pemberian antibiotik sebelum penderita menjalani perawatan gigi atau prosedur bedah minor lainnya, bisa membantu mengurangi resiko terjadinya endokarditis.
Jika PDA telah menutup, baik melalui pembedahan maupun teknik transkateter, maka 6 bulan setelah penutupan ini, tidak perlu lagi diberikan antibiotik sebagai tindakan pencegahan terhadap terjadinya endokarditis.
Duktus arteriosus adalah suatu pembuluh darah yang menghubungkan aorta (pembuluh arteri besar yang mengangkut darah ke seluruh tubuh) dengan arteri pulmonalis (arteri yang membawa darah ke paru-paru), yang merupakan bagian dari peredaran darah yang normal pada janin.
Duktus arteriosus memungkinkan darah untuk tidak melewati paru-paru. Pada janin, fungsi ini penting karena janin tidak menghirup udara sehingga darah janin tidak perlu beredar melewati paru-paru agar mengandung banyak oksigen. Janin menerima oksigen dan zat makanan dari plasenta (ari-ari).
Tetapi pada saat lahir, ketika bayi mulai bernafas, duktus arteriosus akan menutup karena darah harus mengalir ke paru-paru agar mengandung banyak oksigen. Pada 95% bayi baru lahir, penutupan duktus terjadi dalam waktu 48-72 jam.
Kelainan ini bisa terjadi baik pada bayi prematur maupun pada bayi cukup umur, dan ditemukan pada 1 diantara 2500-5000 bayi.
Biasanya gejalanya ringan, tetapi akan semakin berat jika tidak diobati/diperbaiki pada usia 2 tahun.
PENYEBAB
Duktus arteriosus adalah suatu pembuluh darah yang dilapisi oleh otot dan memiliki fungsi khusus. Jika kadar oksigen di dalam darah meningkat (biasanya terjadi segera setelah bayi lahir), otot ini akan mengkerut sehingga duktus menutup.
Pada saat duktus menutup, darah dari jantung bagian kanan hanya mengalir ke paru-paru (seperti yang terjadi pada orang dewasa).
Pada beberapa anak, duktus tidak menutup atau hanya menutup sebagian. Hal ini terjadi karena tidak adanya sensor oksigen yang normal pada otot duktus atau karena kelemahan pada otot duktus.
Adapun faktor resiko terjadinya PDA adalah prematuritas dan sindroma gawat pernafasan.
GEJALA
Jika duktus tetap terbuka, darah yang seharusnya mengalir ke seluruh tubuh akan kembali ke paru-paru sehingga memenuhi pembuluh paru-paru.
Jumlah darah tambahan yang sampai ke paru-paru tergantung kepada ukuran PDA. Jika PDA sangat kecil, maka darah yang melewati PDA hanya sedikit. Pada keadaan ini, anak tidak memiliki gejala sama sekali dan tampak baik-baik saja.
PDA yang kecil dapat diketahui jika pada pemeriksaan dengan stetoskop terdengar murmur (suatu bunyi jantung ekstra yang terderngar jika darah menyembur melalui lubang yang sempit). Semakin kecil lubangnya, maka semakin sedikit darah yang mengalir dan semakin halus bunyi murmur yang terdengar.
Jika PDA memiliki lubang yang besar, maka darah dalam jumlah yang besar akan membanjiri paru-paru. Anak tampak sakit, dengan gejala berupa:
- tidak mau menyusu
- berat badannya tidak bertambah
- berkeringat
- kesulitan dalam bernafas
- denyut jantung yang cepat.
Timbulnya gejala tersebut menunjukkan telah terjadinya gagal jantung kongestif, yang seringkali terjadi pada bayi prematur.
Anak dengan PDA yang kecil tidak memiliki resiko menderita gagal jantung kongestif, tetapi tetap memiliki resiko terjadinya endokarditis.
Endokarditis adalah infeksi pada jantung, katup jantung maupun pembuluh darah jantung. Infeksi ini bisa berakibat fatal dan dapat menyebabkan kematian, stroke serta kelainan fungsi jantung.
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik.
Pada pemeriksaan dengan stetoskop seringkali terdengar murmur.
Pemeriksaan yang biasa dilakukan:
# Rontgen dada
# EKG
# Ekokardiogram.
PENGOBATAN
Jika pada saat bayi berusia beberapa minggu terjadi gagal jantung, maka segera dilakukan pembedahan.
Jika gejalanya hanya berupa murmur, maka pembedahan biasanya dilakukan pada saat anak berusia 1 tahun.
Jika tidak ada gejala, pembedahan ditunda sampai anak berumur 6 bulan-3 tahun.
Terdapat beberapa cara untuk mengatasi PDA, yang pemilihannya tergantung kepada berbagai faktor dan yang terpenting adalah usia anak:
# Bayi prematur
- Pemberian indometasin, yang secara tidak langsung akan merangsang kontraksi (pengkerutan) otot duktus.
Efek samping dari indometasin adalah perdarahan internal dan kelainan fungsi ginjal.
Jika pemberian pertama tidak berhasil dan tidak terjadi efek samping, pemberian indometasin bisa diulang.
- Pembedahan
Jika indometasin tidak efektif atau tidak dapat diberikan karena ada masalah medis lainnya, maka dilakukan pembedahan. Kedua ujung PDA diikat atau dijahit, lalu duktus dipotong.
# Bayi dan anak-anak
Pada bayi cukup umur atau anak-anak yang lebih besar, jarang terjadi gagal jantung kongestif.
Pada bayi berusia dibawah 6 bulan yang menunjukkan tanda-tanda gagal jantung, biasanya dilakukan pembedahan.
Pada anak yang berumur lebih dari 6 bulan dan tidak menunjukkan gejala, dilakukan teknik penutupan transkateter.
Pemberian antibiotik sebelum penderita menjalani perawatan gigi atau prosedur bedah minor lainnya, bisa membantu mengurangi resiko terjadinya endokarditis.
Jika PDA telah menutup, baik melalui pembedahan maupun teknik transkateter, maka 6 bulan setelah penutupan ini, tidak perlu lagi diberikan antibiotik sebagai tindakan pencegahan terhadap terjadinya endokarditis.
Stenosis Katup Aorta
Stenosis Katup Aorta adalah suatu penyempitan atau penyumbatan pada katup aorta.
Katup aorta adalah katup pada ventrikel kiri jantung yang akan membuka ketika darah akan masuk ke dalam aorta lalu diedarkan ke seluruh tubuh.
Dalam keadaan normal, katup aorta terdiri dari 3 kuncup yang akan menutup dan membuka sehingga darah bisa melewatinya.
Pada stenosis katup aorta, biasanya katup hanya terdiri dari 2 kuncup sehingga lubangnya lebih sempit dan bisa menghambat aliran darah. Akibatnya ventrikel kiri harus memompa lebih kuat agar darah bisa melewati katup aorta.
PENYEBAB
Stenosis katup aorta merupakan suatu kelainan jantung bawaan.
GEJALA
Pada beberapa anak terjadi gagal jantung dan penurunan aliran darah ke seluruh tubuh.
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik.
Pada pemeriksaan dengan stetoskop mungkin akan terdengar murmur (bunyi jantung abnormal yang terjadi jika darah melewati saluran yang sempit).
Pemeriksaan yang biasa dilakukan:
- Ekokardiogram
- USG Doppler
- Kateterisasi jantung kiri
- Rontgen dada
- EKG.
PENGOBATAN
Pengobatan biasanya terdiri dari obat-obatan dan pembedahan darurat atau valvoplasti balon (peregangan katup dengan menggunakan selang yang pada ujungnya terpasang balon).
Pada anak-anak yang lebih besar, bisa dilakukan pembedahan untuk membuka katup atau menggantinya dengan katup buatan.
Katup aorta adalah katup pada ventrikel kiri jantung yang akan membuka ketika darah akan masuk ke dalam aorta lalu diedarkan ke seluruh tubuh.
Dalam keadaan normal, katup aorta terdiri dari 3 kuncup yang akan menutup dan membuka sehingga darah bisa melewatinya.
Pada stenosis katup aorta, biasanya katup hanya terdiri dari 2 kuncup sehingga lubangnya lebih sempit dan bisa menghambat aliran darah. Akibatnya ventrikel kiri harus memompa lebih kuat agar darah bisa melewati katup aorta.
PENYEBAB
Stenosis katup aorta merupakan suatu kelainan jantung bawaan.
GEJALA
Pada beberapa anak terjadi gagal jantung dan penurunan aliran darah ke seluruh tubuh.
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik.
Pada pemeriksaan dengan stetoskop mungkin akan terdengar murmur (bunyi jantung abnormal yang terjadi jika darah melewati saluran yang sempit).
Pemeriksaan yang biasa dilakukan:
- Ekokardiogram
- USG Doppler
- Kateterisasi jantung kiri
- Rontgen dada
- EKG.
PENGOBATAN
Pengobatan biasanya terdiri dari obat-obatan dan pembedahan darurat atau valvoplasti balon (peregangan katup dengan menggunakan selang yang pada ujungnya terpasang balon).
Pada anak-anak yang lebih besar, bisa dilakukan pembedahan untuk membuka katup atau menggantinya dengan katup buatan.
Koartasio Aorta
Aorta adalah arteri utama pada tubuh. Aorta mengedarkan darah yang kaya akan oksigen ke seluruh bagian tubuh, kecuali paru-paru.
Cabang pertama dari aorta mengalirkan darah ke tubuh bagian atas (lengan dan kepala). Kemudian darah mengalir ke tubuh bagian bawah (perut dan tungkai).
PENYEBAB
Resiko terjadinya koartasio aorta meningkat pada beberapa keadaan genetik, seperti sindroma Turner.
Koartasio aorta juga berhubungan dengan kelainan bawaan pada katup aorta (misalnya katup bikuspidalis).
Kelainan ini ditemukan pada 1 dari 10.000 orang. Biasanya terdiagnosis pada masa kanak-kanak atau dewasa dibawah 40 tahun.
GEJALA
Gejalanya mungkin baru timbul pada masa remaja, tetapi bisa juga muncul pada saat bayi, tergantung kepada beratnya tahanan terhadap aliran darah.
Gejalanya berupa:
- pusing
- pingsan
- kram tungkai pada saat melakukan aktivitas
- tekanan darah tinggi yang terlokalisir (hanya pada tubuh bagian atas)
- kaki atau tungkai teraba dingin
- kekurangan tenaga
- sakit kepala berdenyut
- perdarahan hidung
- nyeri tungkai selama melakukan aktivitas.
Pada usia beberapa hari sampai 2 minggu, setelah duktus ateriosus menutup, beberapa bayi mengalami gagal jantung. Terjadi gangguan pernafasan yang berat, bayi tampak sangat pucat dan pemeriksaan darah menunjukkan peningkatan asam di dalam darah (asidosis metabolik).

DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik yang menunjukkan:
- tekanan darah tinggi dilengan, dengan perbedaan tekanan yang signifikan antara lengan dan tungkai
- denyut nadi femoralis (selangkangan) lebih lemah dibandingkan dengan denyut nadi karotis (leher) atau denyut nadi femoralis sama sekali tak teraba
- dengan bantuan stetoskop bisa terdengar murmur (bunyi jantung abnormal)
- mungkin ditemukan tanda-tanda gagal jantung kiri (terutama pada bayi) atau tanda-tanda dari regurgitasi aorta.
Untuk memperkuat diagnosis, dilakukan pemeriksaan berikut:
# Angiografi koroner
# CT scan dada
# MRI dada
# Ekokardiografi
# USG Doppler aorta
# Rontgen dada
# EKG (menunjukkan adanya pembesaran ventrikel kiri)
# Kateterisasi jantung.
PENGOBATAN
Kelainan ini sebaiknya segera diperbaiki pada awal masa kanak-kanak untuk mengurangi beban kerja pada ventrikel kiri. Pembedahan biasanya dilakukan pada usia prasekolah (biasanya umur 3-5 tahun).
Jika terjadi gagal jantung, segera diberikan prostaglandin untuk membuka duktus arteriosus dan obat lainnya untuk memperkuat jantung serta pembedahan darurat untuk memperbaiki aorta.
Bagian aorta yang menyempit dapat dibuang melalui pembedahan atau kadang dilakukan tindakan non-bedah berupa kateterisasi balon untuk melebarkan bagian yang menyempit.
Pada pembedahan, bagian aorta yang menyempit dibuang. Jika bagian yang terbuang hanya sedikit, kemudian dibuat anastomisis (penyambungan kembali kedua ujung aorta) atau kedua ujung aorta dijembatani oleh pencangkokan dakron.
Kekambuhan koartasio aorta jarang terjadi jika:
- pembedahan dilakukan pada masa bayi atau masa kanak-kanak
- sampai masa dewasa tidak ditemukan perbedaan tekanan darah antara lengan dan tungkai.
Koartasio kambuhan biasanya diatasi dengan pelebaran balon non-bedah atau dengan pencangkokan suatu bahan melalui prosedur kateterisasi.
Transposisi Arteri Besar
Transposisi Arteri Besar adalah kelainan letak dari aorta dan arteri pulmonalis.
Dalam keadaan normal, aorta berhubungan dengan ventrikel kiri jantung dan arteri pulmonalis berhubungan dengan ventrikel kanan jantung. Pada transposisi arteri besar yang terjadi adalah kebalikannya. Aorta terletak di ventikel kanan jantung dan arteri pulmonalis terletak di ventrikel kiri jantung.
Darah dari seluruh tubuh yang kekurangan oksigen akan mengalir ke dalam aorta dan kembali dialirkan ke seluruh tubuh. Sedangkan darah yang berasal dari paru-paru dan kaya akan oksigen akan kembali dialirkan ke paru-paru.
Transposisi arteri besar dikelompokkan ke dalam kelainan jantung sianotik, dimana terjadi pemompaan darah yang kekurangan oksigen ke seluruh tubuh, yang menyebabkan sianosis (kulit menjadi ungu kebiruan) dan sesak nafas.
Bayi dengan kelainan ini, setelah lahir bisa bertahan sebentar saja karena adanya lubang diantara atrium kiri dan kanan yang disebut foramen ovale.
Foramen ovale ini dalam keadaan normal ditemukan pada bayi ketika lahir. Dengan adanya lubang ini, maka sejumlah kecil darah yang kaya akan oksigen akan mengalir dari atrium kiri ke atrium kanan, lalu ke ventrikel kanan dan ke aorta sehingga mampu memenuhi kebutuhan tubuh akan oksigen dan bayi tetap hidup.

PENYEBAB
Penyebab dari kebanyakan kelainan jantung bawaan tidak diketahui.
Faktor-faktor prenatal (sebelum bayi lahir) yang berhubungan dengan transposisi arteri besar adalah:
- Rubella (campak Jerman) atau infeksi virus lainnya pada ibu hamil
- Nutrisi yang buruk selama kehamilan
- Ibu yang alkoholik
- Usia ibu lebih dari 40 tahun
- Ibu menderita diabetes.
Transposisi arteri besar terjadi pada 40 dari 100.000 bayi. Kelainan ini merupakan kelainan jantung sianotik yang paling sering ditemukan pada minggu pertama kehidupan seorang bayi.
GEJALA
Gejalanya berupa:
- sianosis
- sesak nafas
- tidak mau makan/menyusu
- jari tangan atau kaki clubbing (seperti tabuh genderang).

DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik.
Pada pemeriksaan dengan stetoskop akan terdengar murmur (bunyi jantung abnormal).
Pemeriksaan yang biasa dilakukan:
- Rontgen dada
- Kateterisasi jantung
- EKG
- Ekokardiogram.
PENGOBATAN
Untuk memperbaiki transposisi arteri besar biasanya dilakukan pembedahan.
Sebelum pembedahan dilakukan, mungkin perlu diberikan prostaglandin agar duktus arteriosus tetap terbuka.
Pada beberapa bayi perlu dilakukan pelebaran foramen ovale dengan selang yang pada ujungnya terpasang balon, agar darah yang kaya oksigen lebih banyak yang masuk ke aorta.
Terdapat 2 jenis pembedahan utama yang bisa dilakukan untuk memperbaiki transposisi arteri besar:
1. Membuat sebuah terowongan diantara atrium. Dengan cara ini, darah yang kaya oksigen akan mengalir ke ventrikel kanan lalu masuk ke aorta, sedangkan darah yang kekurangan oksigen akan mengalir ke ventrikel kiri dan masuk ke dalam arteri pulmonalis. Pembedahan ini disebut atrial switch atau venous switch, atau prosedur Mustard maupun prosedur Senning.
2. Pembedahan arterial switch. Aorta dan arteri pulmoner dikembalikan ke posisinya yang normal. Aorta dihubungkan dengan ventrikel kiri dan arteri pulmonalis dihubungkan dengan ventrikel kanan.
Arteri koroner yang membawa darah kaya oksigen sebagai sumber energi bagi otot jantung, juga kembali disambungkan dengan aorta yang baru.
Dalam keadaan normal, aorta berhubungan dengan ventrikel kiri jantung dan arteri pulmonalis berhubungan dengan ventrikel kanan jantung. Pada transposisi arteri besar yang terjadi adalah kebalikannya. Aorta terletak di ventikel kanan jantung dan arteri pulmonalis terletak di ventrikel kiri jantung.
Darah dari seluruh tubuh yang kekurangan oksigen akan mengalir ke dalam aorta dan kembali dialirkan ke seluruh tubuh. Sedangkan darah yang berasal dari paru-paru dan kaya akan oksigen akan kembali dialirkan ke paru-paru.
Transposisi arteri besar dikelompokkan ke dalam kelainan jantung sianotik, dimana terjadi pemompaan darah yang kekurangan oksigen ke seluruh tubuh, yang menyebabkan sianosis (kulit menjadi ungu kebiruan) dan sesak nafas.
Bayi dengan kelainan ini, setelah lahir bisa bertahan sebentar saja karena adanya lubang diantara atrium kiri dan kanan yang disebut foramen ovale.
Foramen ovale ini dalam keadaan normal ditemukan pada bayi ketika lahir. Dengan adanya lubang ini, maka sejumlah kecil darah yang kaya akan oksigen akan mengalir dari atrium kiri ke atrium kanan, lalu ke ventrikel kanan dan ke aorta sehingga mampu memenuhi kebutuhan tubuh akan oksigen dan bayi tetap hidup.
PENYEBAB
Penyebab dari kebanyakan kelainan jantung bawaan tidak diketahui.
Faktor-faktor prenatal (sebelum bayi lahir) yang berhubungan dengan transposisi arteri besar adalah:
- Rubella (campak Jerman) atau infeksi virus lainnya pada ibu hamil
- Nutrisi yang buruk selama kehamilan
- Ibu yang alkoholik
- Usia ibu lebih dari 40 tahun
- Ibu menderita diabetes.
Transposisi arteri besar terjadi pada 40 dari 100.000 bayi. Kelainan ini merupakan kelainan jantung sianotik yang paling sering ditemukan pada minggu pertama kehidupan seorang bayi.
GEJALA
Gejalanya berupa:
- sianosis
- sesak nafas
- tidak mau makan/menyusu
- jari tangan atau kaki clubbing (seperti tabuh genderang).
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik.
Pada pemeriksaan dengan stetoskop akan terdengar murmur (bunyi jantung abnormal).
Pemeriksaan yang biasa dilakukan:
- Rontgen dada
- Kateterisasi jantung
- EKG
- Ekokardiogram.
PENGOBATAN
Untuk memperbaiki transposisi arteri besar biasanya dilakukan pembedahan.
Sebelum pembedahan dilakukan, mungkin perlu diberikan prostaglandin agar duktus arteriosus tetap terbuka.
Pada beberapa bayi perlu dilakukan pelebaran foramen ovale dengan selang yang pada ujungnya terpasang balon, agar darah yang kaya oksigen lebih banyak yang masuk ke aorta.
Terdapat 2 jenis pembedahan utama yang bisa dilakukan untuk memperbaiki transposisi arteri besar:
1. Membuat sebuah terowongan diantara atrium. Dengan cara ini, darah yang kaya oksigen akan mengalir ke ventrikel kanan lalu masuk ke aorta, sedangkan darah yang kekurangan oksigen akan mengalir ke ventrikel kiri dan masuk ke dalam arteri pulmonalis. Pembedahan ini disebut atrial switch atau venous switch, atau prosedur Mustard maupun prosedur Senning.
2. Pembedahan arterial switch. Aorta dan arteri pulmoner dikembalikan ke posisinya yang normal. Aorta dihubungkan dengan ventrikel kiri dan arteri pulmonalis dihubungkan dengan ventrikel kanan.
Arteri koroner yang membawa darah kaya oksigen sebagai sumber energi bagi otot jantung, juga kembali disambungkan dengan aorta yang baru.
Sindroma Hipoplastik Jantung Kiri
Sindroma Ventrikel Kiri Yg Tidak
Berkembang (Sindroma Hipoplastik Jantung Kiri) adalah suatu keadaan
dimana bagian kiri jantung tidak berkembang. .
Tugas utama dari ventrikel kiri adalah memompa darah ke seluruh tubuh.
Jika ventrikel kiri dan katupnya tidak berkembang atau tidak terbentuk, maka akan terjadi gangguan aliran darah ke seluruh tubuh.
Jantung yang normal dan cara kerjanya
Jantung yang normal adalah suatu otot pemompa yang kuat, yang ukurannya sedikit lebih besar dari kepalan tinju.
Jantung terus menerus memompa darah melalui sistem peredaran. Setiap hari jantung rata-rata berdenyut sebanyak 100.000 kali dan memompa sekitar 2000 galon darah.
Jantung memiliki 4 ruang, ruang sebelah atas disebut atrium dan terbagi oleh suatu sekat (septum menjadi atrium kiri dan atrium kanan; ruang sebelah bawah disebut ventrikel dan terbagi oleh septum menjadi ventrikel kiri dan ventrikel kanan.
Pemompaan darah melalui keempat ruang tersebut dibantu oleh 4 katup jantung. Katup membuka dan menutup sehingga darah hanya mengalir dalam satu arah.
Keempat katup jantung tersebut adalah:
1. Katup trikuspidalis, terletak diantara atrium kanan dan ventrikel kanan
2. Katup pulmonalis, terletak diantara ventrikel kanan dan arteri pulmonalis
3. Katup mitralis, terletak diantara atrium kiri dan ventrikel kiri
4. Katup aorta, terletak diantara ventrikel kiri dan aorta.
Setiap katup memiliki sejumlah daun. Katup mitralis memiliki 2 daun, sedangkan yang lainnya memiliki 3 daun.

Darah berwarna gelap kebiruan adalah darah yang kekurangan oksigen, yang mengalir kembali ke jantung setelah beredar ke seluruh tubuh. Darah ini kembali ke jantung melalui vena dan masuk ke atrium kanan. Melalui katup trikuspidalis, darah akan mengalir ke ventrikel kanan.
Ventrikel kanan memompa darah melalui katup pulmonalis dan masuk ke dalam arteri pulmonalis. Dari arteri pulmonalis, darah masuk ke paru-paru dan mendapatkan oksigen yang segar. Setelah mendapatkan oksigen yang segar, warna darah menjadi merah terang.
Dari paru-paru, darah masuk ke dalam atrium kiri melalui vena pulmonalis. Dari atrium kiri darah masuk ke ventrikel kiri melalui katup mitralis.
Ventrikel kiri memompa darah yang kaya akan oksigen melalui katup aorta ke dalam aorta. Aorta mengangkut darah ke seluruh tubuh.
Tekanan darah di dalam ventrikel kiri sama dengan tekanan darah yang terukur di lengan.
PENYEBAB
Sindroma hipoplastik jantung kiri adalah suatu kelainan jantung bawaan.
Hal ini terjadi jika terdapat gangguan pada perkembangan ventrikel kiri dan struktur pembuluh darah yang berhubungan dengannya (katup mitralis, katup aorta dan pulmonalis).
Jantung bagian kiri tidak mampu mempertahankan peredaran darah ke seluruh tubuh. Karena jantung kiri tidak dapat berfungsi, maka perdaran darah pulmoner (paru-paru) dan sistemik (seluruh tubuh) harus dijalankan oleh jantung bagian kanan. Hal ini akan menyebabkan terjadinya gagal jantung kanan.
Satu-satunya kemungkinan bagi bayi untuk bertahan hidup adalah adanya hubungan antara peredaran darah kiri dan kanan yang disebut shunt. Semua bayi yang normal terlahir dengan 2 shunt, yaitu foramen ovale dan duktus arteriosus, yang beberapa hari setelah lahir akan menutup secara spontan.
GEJALA
Ketika lahir, bayi tampak normal. Gejala timbul secara samar dan pada awalnya bersifat ringan.
Gejalanya berupa:
- bayi tampak lemas
- tidak mau menyusu atau makan
- sesak nafas
- warna kulitnya pucat atau kebiruan (sianosis).
Pada bayi baru lahir yang sehat, warna kebiruan muncul jika bayi kedinginan dan terlihat pada tangan, kaki dan wajahnya (keadaan ini disebut sianosis perifer)
Jika warna kebiruan tampak pada dada atau perut, bibir dan lidah, maka disebut sianosis sentral. Keadaan ini adalah abnormal karena menunjukkan adanya kekurangan oksigen di dalam darah arteri yang merupakan akibat dari kelainan jantung dan peredaran darah. Jika bayi menangis, maka sianosis sentral semakin meningkat dan tidak berkurang meskipun bayi diberi kehangatan.
Pada saat lahir, bayi tampak normal karena darah dapat mengalir dari ventrikel kanan ke seluruh tubuh melalui duktus arteriosus yang masih membuka. Tetapi ketika duktus telah menutup, akan terjadi gagal jantung yang berat.
Kebanyakan bayi yang menderita sindroma ini akan meninggal.
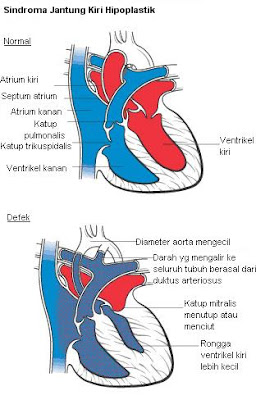
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik, yang menunjukkan adanya gagal jantung kongestif, yaitu hepatomegali (pembesaran hati). Selain itu, denyut nadi pada berbagai lokasi (pergelangan tangan, selangkangan dan lainnya) teraba lemah.
Pemeriksaan yang biasa dilakukan:
# EKG (tampak adanya pembesaran ventrikel kanan)
# Rontgen dada (menunjukkan adanya pembesaran jantung)
# Ekokardiogram (menunjukkan adanya hipoplastik pada ventrikel kiri)
# Kateterisasi jantung (pada beberapa kasus perlu dilakukan untuk melengkapi ekokardiogram).
PENGOBATAN
Sebelum pembedahan dilakukan, langkah pertama yang harus dilakukan adalah memberikan obat prostaglandin, yang membantu agar duktus arteriosus tetap membuka sehingga peredaran darah bisa tetap terjaga.
Keadaan ini bisa disembuhkan dengan pencangkokan jantung, tetapi anak harus minum obat seumur hidupnya guna mencegah penolakan tubuh terhadap jantung yang dicangkokkan.
Beberapa tindakan paliatif yang bisa meringankan gejala:
# Oksigen
# Obat-obatan : PGE1 (prostaglandin), dopamin, kalsium 3
# Prosedur Norwood
- Tahap I : membuat hubungan antara peredaran darah sistemik dengan pulmoner (misalnya antara aorta dengan arteri pulmonalis) dan melebarkan aorta. Tahap I perlu diikuti dengan pembedahan Fontan atau pencangkokan jantung.
- Tahap II dan III : membuat hubungan yang disebut shunt kavopulmoner. Selama tahap II (prosedur hemi-Fontan), vena kava superior dihubungkan dengan arteri pulmonalis. Selama tahap III, vena kava inferior dihubungkan dengan ventrikel (prosedur Fontan komplit). Tahap terakhir ini biasanya dilakukan pada saat anak berumur 12-18 bulan.
Prosedur Norwood merupakan pilihan pengobatan terbaik jika tidak mungkin dilakukan pencangkokan jantung.
# Jika prosedur Norwood tahap I telah berhasil dilakukan, bisa dilakukan pencangkokan jantung.
Tetralogi Fallot
- Defek septum ventrikel (lubang diantara ventrikel kiri dan kanan)
- Stenosis katup pulmoner (penyempitan pada katup pulmonalis)
- Transposisi aorta
- Hipertrofi ventrikel kanan (penebalan otot ventrikel kanan).

PENYEBAB
Kebanyakan penyebab dari kelainan jantung bawaan tidak diketahui. Biasanya melibatkan berbagai faktor.
Faktor prenatal yang berhubungan dengan resiko terjadinya tetralogi Fallot adalah:
- Selama hamil, ibu menderita rubella (campak Jerman) atau infeksi virus lainnya
- Gizi yang buruk selama hamil
- Ibu yang alkoholik
- Usia ibu diatas 40 tahun
- Ibu menderita diabetes.
Tetralogi Fallot lebih sering ditemukan pada anak-anak yang menderita sindroma Down.
Tetralogi Fallot dimasukkan ke dalam kelainan jantung sianotik karena terjadi pemompaan darah yang sedikit mengandung oksigen ke seluruh tubuh, sehingga terjadi sianosis (kulit berwarna ungu kebiruan) dan sesak nafas.
Mungkin gejala sianotik baru timbul di kemudian hari, dimana bayi mengalami serangan sianotik karena menyusu atau menangis.
Tetralogi Fallot terjadi pada sekitar 50 dari 100.000 bayi dan merupakan kelainan jantung bawaan nomor 2 yang paling sering terjadi.
GEJALA
Gejalanya bisa berupa:
- bayi mengalami kesulitan untuk menyusu
- berat badan bayi tidak bertambah
- pertumbuhan anak berlangsung lambat
- perkembangan anak yang buruk
- sianosis
- jari tangan clubbing (seperti tabuh genderang karena kulit atau tulang di sekitar kuku jari tangan membesar)
- sesak nafas jika melakukan aktivitas
- setelah melakukan aktivitas, anak selalu jongkok.
Serangan sianosis biasanya terjadi ketika anak melakukan aktivitas (misalnya menangis atau mengedan), dimana tiba-tiba sianosis memburuk sehingga anak menjadi sangat biru, mengalami sesak nafas dan bisa pingsan.
DIAGNOSA
Pada pemeriksaan dengan stetoskop biasanya akan terdengar murmur (bunyi jantung yang abnormal).
Pemeriksaan yang biasa dilakukan:
- EKG
- Pemeriksaan darah lengkap menunjukkan adanya peningkatan jumlah sel darah merah dan hematokrit
- Rontgen dada menunjukkan ukuran hati yang kecil
- Kateterisasi jantung
- Ekokardiogram.
PENGOBATAN
Pada serangan sianosis, diberikan oksigen dan morfin. Untuk mencegah serangan lainnya, untuk sementara waktu bisa diberikan propanolol.
Pembedahan untuk memperbaiki kelainan jantung ini biasanya dilakukan ketika anak berumur 3-5 tahun (usia pra-sekolah).
Pada kelainan yang lebih berat, pembedahan bisa dilakukan lebih awal.
Pembedahan yang dilakukan terdiri dari 2 tahap:
1. Pembedahan sementara
Pembuatan shunt bisa terlebih dahulu dilakukan pada bayi yang kecil dan sangat biru, agar aliran darah ke paru-paru cukup. Shunt dibuat diantara aorta dan arteri pulmonalis.
Setelah bayi tumbuh cukup besar, dilakukan pembedahan perbaikan untuk menutup kembali shunt tersebut.
2. Pembedahan perbaikan terdiri dari:
- penutupan VSD
- pembukaan jalur aliran ventrikel kanan dengan cara membuang sebagian otot yang berada di bawah katup pulmonalis
- perbaikan atau pengangkatan katup pulmonalis
- pelebaran arteri pulmonalis perifer yang menuju ke paru-paru kiri dan kanan.
Kadang diantara ventrikel kanan dan arteri pulmonalis dipasang sebuah selang (perbaikan Rastelli).
Jika tidak dilakukan pembedahan, penderita biasanya akan meninggal pada usia 20 tahun.
Orang tua dari anak-anak yang menderita kelainan jantung bawaan bisa diajari tentang cara-cara menghadapi gejala yang timbul:
# Menyusui atau menyuapi anak secara perlahan
# Memberikan porsi makan yang lebih kecil tetapi lebih sering
# Mengurangi kecemasan anak dengan tetap bersikap tenang
# Menghentikan tangis anak dengan cara memenuhi kebutuhannya
# Membaringkan anak dalam posisi miring dan kaki ditekuk ke dada selama serangan sianosis.
Atresia Esofagus
Atresia Esofagus adalah esofagus (kerongkongan) yang tidak terbentuk secara sempurna.
Pada atresi esofagus, kerongkongan menyempit atau buntu; tidak tersambung dengan lambung sebagaimana mestinya.
Kebanyakan bayi yang menderita atresia esofagus juga memiliki fistula trakeoesofageal (suatu hubungan abnormal antara kerongkongan dan trakea/pipa udara).

PENYEBAB
Atresia esofagus merupakan suatu kelainan bawaan pada saluran pencernaan.
Terdapat beberapa jenis atresia, tetapi yang paling sering ditemukan adalah kerongkongan yang buntu dan tidak tersambung dengan kerongkongan bagian bawah serta lambung.
Atresia esofagus dan fistula ditemuka pada 2-3 dari 10.000 bayi.
GEJALA
Gejalanya bisa berupa:
- mengeluarkan ludah yang sangat banyak
- terbatuk atau tersedak setelah berusaha untuk menelan
- tidak mau menyusu
- sianosis (kulitnya kebiruan).
Adanya fistula menyebabkan ludah bisa masuk ke dalam paru-paru sehingga terdapat resiko terjadinya pneumonia aspirasi.
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan berikut:
# Memasukkan selang nasogastrik
# Rontgen esofagus menunjukkan adanya kantong udara dan adanya udara di lambung serta usus.
PENGOBATAN
Jika keadaan bayi stabil, dilakukan pembedahan untuk memperbaiki atresia dan menutup fistula.
Sebelum pembedahan dilakukan, untuk mencegah pneumonia aspirasi, makanan bayi diberikan melalui infus dan pada kerongkongan bagian atas dipasang alat penghisap ludah agar tidak masuk ke paru-paru.
Pada atresi esofagus, kerongkongan menyempit atau buntu; tidak tersambung dengan lambung sebagaimana mestinya.
Kebanyakan bayi yang menderita atresia esofagus juga memiliki fistula trakeoesofageal (suatu hubungan abnormal antara kerongkongan dan trakea/pipa udara).
PENYEBAB
Atresia esofagus merupakan suatu kelainan bawaan pada saluran pencernaan.
Terdapat beberapa jenis atresia, tetapi yang paling sering ditemukan adalah kerongkongan yang buntu dan tidak tersambung dengan kerongkongan bagian bawah serta lambung.
Atresia esofagus dan fistula ditemuka pada 2-3 dari 10.000 bayi.
GEJALA
Gejalanya bisa berupa:
- mengeluarkan ludah yang sangat banyak
- terbatuk atau tersedak setelah berusaha untuk menelan
- tidak mau menyusu
- sianosis (kulitnya kebiruan).
Adanya fistula menyebabkan ludah bisa masuk ke dalam paru-paru sehingga terdapat resiko terjadinya pneumonia aspirasi.
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan berikut:
# Memasukkan selang nasogastrik
# Rontgen esofagus menunjukkan adanya kantong udara dan adanya udara di lambung serta usus.
PENGOBATAN
Jika keadaan bayi stabil, dilakukan pembedahan untuk memperbaiki atresia dan menutup fistula.
Sebelum pembedahan dilakukan, untuk mencegah pneumonia aspirasi, makanan bayi diberikan melalui infus dan pada kerongkongan bagian atas dipasang alat penghisap ludah agar tidak masuk ke paru-paru.
Penyakit Hirschprung
Penyakit Hirschsprung (Megakolon
Kongenital) adalah suatu penyumbatan pada usus besar yang terjadi akibat
pergerakan usus yang tidak adekuat karena sebagian dari usus besar
tidak memiliki saraf yang mengendalikan kontraksi ototnya.

PENYEBAB
Dalam keadaan normal, bahan makanan yang dicerna bisa berjalan di sepanjang usus karena adanya kontraksi ritmis dari otot-otot yang melapisi usus (kontraksi ritmis ini disebut gerakan peristaltik).
Kontraksi otot-otot tersebut dirangsang oleh sekumpulan saraf yang disebut ganglion, yang terletak dibawah lapisan otot.
Pada penyakit Hirschsprung, ganglion ini tidak ada, biasanya hanya sepanjang beberapa sentimeter.
Segmen usus yang tidak memiliki gerakan peristaltik tidak dapat mendorong bahan-bahan yang dicerna dan terjadi penyumbatan.
Penyakit Hirschsprung 5 kali lebih sering ditemukan pada bayi laki-laki.
Penyakit ini kadang disertai dengan kelainan bawaan lainnya, misalnya sindroma Down.
GEJALA
Gejala-gejala yang mungkin terjadi:
- segera setelah lahir, bayi tidak dapat mengeluarkan mekonium (tinja pertama pada bayi baru lahir)
- tidak dapat buang air besar dalam waktu 24-48 jam setelah lahir
- perut menggembung
- muntah
- diare encer (pada bayi baru lahir)
- berat badan tidak bertambah
- malabsorbsi.
Kasus yang lebih ringan mungkin baru akan terdiagnosis di kemudian hari.
Pada anak yang lebih besar, gejalanya adalah sembelit menahun, perut menggembung dan gangguan pertumbuhan.
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik.
Pemeriksaan colok dubur (memasukkan jari tangan ke dalam anus) menunjukkan adanya pengenduran pada otot rektum.
Pemeriksaan yang biasa dilakukan:
# Rontgen perut (menunjukkan pelebaran usus besar yang terisi oleh gas dan tinja)
# Barium enema
# Manometri anus (pengukuran tekanan sfingter anus dengan cara mengembangkan balon di dalam rektum)
# Biopsi rektum (menunjukkan tidak adanya ganglion sel-sel saraf).
PENGOBATAN
Untuk mencegah terjadinya komplikasi akibat penyumbatan usus, segera dilakukan kolostomi sementara. Kolostomi adalah pembuatan lubang pada dinding perut yang disambungkan dengan ujung usus besar.
Pengangkatan bagian usus yang terkena dan penyambungan kembali usus besar biasanya dilakukan pada saat anak berusia 6 bulan atau lebih.
Jika terjadi perforasi (perlubangan usus) atau enterokolitis, diberikan antibiotik.
PENYEBAB
Dalam keadaan normal, bahan makanan yang dicerna bisa berjalan di sepanjang usus karena adanya kontraksi ritmis dari otot-otot yang melapisi usus (kontraksi ritmis ini disebut gerakan peristaltik).
Kontraksi otot-otot tersebut dirangsang oleh sekumpulan saraf yang disebut ganglion, yang terletak dibawah lapisan otot.
Pada penyakit Hirschsprung, ganglion ini tidak ada, biasanya hanya sepanjang beberapa sentimeter.
Segmen usus yang tidak memiliki gerakan peristaltik tidak dapat mendorong bahan-bahan yang dicerna dan terjadi penyumbatan.
Penyakit Hirschsprung 5 kali lebih sering ditemukan pada bayi laki-laki.
Penyakit ini kadang disertai dengan kelainan bawaan lainnya, misalnya sindroma Down.
GEJALA
Gejala-gejala yang mungkin terjadi:
- segera setelah lahir, bayi tidak dapat mengeluarkan mekonium (tinja pertama pada bayi baru lahir)
- tidak dapat buang air besar dalam waktu 24-48 jam setelah lahir
- perut menggembung
- muntah
- diare encer (pada bayi baru lahir)
- berat badan tidak bertambah
- malabsorbsi.
Kasus yang lebih ringan mungkin baru akan terdiagnosis di kemudian hari.
Pada anak yang lebih besar, gejalanya adalah sembelit menahun, perut menggembung dan gangguan pertumbuhan.
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik.
Pemeriksaan colok dubur (memasukkan jari tangan ke dalam anus) menunjukkan adanya pengenduran pada otot rektum.
Pemeriksaan yang biasa dilakukan:
# Rontgen perut (menunjukkan pelebaran usus besar yang terisi oleh gas dan tinja)
# Barium enema
# Manometri anus (pengukuran tekanan sfingter anus dengan cara mengembangkan balon di dalam rektum)
# Biopsi rektum (menunjukkan tidak adanya ganglion sel-sel saraf).
PENGOBATAN
Untuk mencegah terjadinya komplikasi akibat penyumbatan usus, segera dilakukan kolostomi sementara. Kolostomi adalah pembuatan lubang pada dinding perut yang disambungkan dengan ujung usus besar.
Pengangkatan bagian usus yang terkena dan penyambungan kembali usus besar biasanya dilakukan pada saat anak berusia 6 bulan atau lebih.
Jika terjadi perforasi (perlubangan usus) atau enterokolitis, diberikan antibiotik.
Omfalokel
Omfalokel terjadi pada 1 dari 5.000 kelahiran.
Usus terlihat dari luar melalui selaput peritoneum yang tipis dan transparan (tembus pandang).

PENYEBAB
Penyebabnya tidak diketahui.
Pada 25-40% bayi yang menderita omfalokel, kelainan ini disertai oleh kelainan bawaan lainnya, seperti kelainan kromosom, hernia diafragmatika dan kelainan jantung.
GEJALA
Banyaknya usus dan organ perut lainnya yang menonjol pada omfalokel bervariasi, tergantung kepada besarnya lubang di pusar.
Jika lubangnya kecil, mungkin hanya usus yang menonjol; tetapi jika lubangnya besar, hati juga bisa menonjol melalui lubang tersebut.
DIAGNOSA
Diagnosis ditegakkan berdasarkan hasil pemeriksaan fisik, dimana isi perut terlihat dari luar melalui selaput peritoneum.
PENGOBATAN
Agar tidak terjadi cedera pada usus dan infeksi perut, segera dilakukan pembedahan untuk menutup omfalokel.
Atresia Anus : Lubang Anus Tidak Terbentuk
Atresia Anus (Anus Imperforatus) adalah suatu keadaan dimana lubang anus tidak terbentuk.
Kebanyakan bayi yang menderita atresia anus juga memiliki fistula (hubungan abnormal) antara anus dengan uretra, perineum maupun kandung kemih.

PENYEBAB
Atresia anus adalah suatu kelainan bawaan.
Kelainan ini ditemukan pada 1 diantara 5000 bayi.
GEJALA
Gejalanya adalah:
- mekonium (tinja pertama pada bayi baru lahir) tidak keluar dalam waktu 24-48 jam setelah lahir
- tinja keluar dari vagina atau uretra
- perut menggembung
- jika disusui, bayi akan muntah .
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik.
PENGOBATAN
Dilakukan pembedahan untuk membentuk lubang anus
Kebanyakan bayi yang menderita atresia anus juga memiliki fistula (hubungan abnormal) antara anus dengan uretra, perineum maupun kandung kemih.
PENYEBAB
Atresia anus adalah suatu kelainan bawaan.
Kelainan ini ditemukan pada 1 diantara 5000 bayi.
GEJALA
Gejalanya adalah:
- mekonium (tinja pertama pada bayi baru lahir) tidak keluar dalam waktu 24-48 jam setelah lahir
- tinja keluar dari vagina atau uretra
- perut menggembung
- jika disusui, bayi akan muntah .
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik.
PENGOBATAN
Dilakukan pembedahan untuk membentuk lubang anus
Atresia Bilier
Fungsi dari sistem empedu adalah membuang limbah metabolik dari hati dan mengangkut garam empedu yang diperlukan untuk mencerna lemak di dalam usus halus.
Pada atresia bilier terjadi penyumbatan aliran empedu dari hati ke kandung empedu. Hal ini bisa menyebabkan kerusakan hati dan sirosis hati, yang jika tidak diobati bisa berakibat fatal.
PENYEBAB
Atresia bilier terjadi karena adanya perkembangan abnormal dari saluran empedu di dalam maupun diluar hati. Tetapi penyebab terjadinya gangguan perkembangan saluran empedu ini tidak diketahui.
Atresia bilier ditemukan pada 1 dari 15.000 kelahiran.
GEJALA
Gejala biasanya timbul dalam waktu 2 minggu setelah lahir, yaitu berupa:
- air kemih bayi berwarna gelap
- tinja berwarna pucat
- kulit berwarna kuning
- berat badan tidak bertambah atau penambahan berat badan berlangsung lambat
- hati membesar.
Pada saat usia bayi mencapai 2-3 bulan, akan timbul gejala berikut:
- gangguan pertumbuhan
- gatal-gatal
- rewel
- tekanan darah tinggi pada vena porta (pembuluh darah yang mengangkut darah dari lambung, usus dan limpa ke hati).
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik.
Pada pemeriksaan perut, hati teraba membesar.
Pemeriksaan yang biasa dilakukan:
# Pemeriksaan darah (terdapat peningkatan kadar bilirubin)
# USG perut
# Rontgen perut (tampak hati membesar)
# Kolangiogram
# Biopsi hati
# Laparotomi (biasanya dilakukan sebelum bayi berumur 2 bulan).
PENGOBATAN
Prosedur yang terbaik adalah mengganti saluran empedu yang mengalirkan empedu ke usus. Tetapi prosedur ini hanya mungkin dilakukan pada 5-10% penderita.
Untuk melompati atresia bilier dan langsung menghubungkan hati dengan usus halus, dilakukan pembedahan yang disebut prosedur Kasai.
Pembedahan akan berhasil jika dilakukan sebelum bayi berusia 8 minggu.
Biasanya pembedahan ini hanya merupakan pengobatan sementara dan pada akhirnya perlu dilakukan pencangkokan hati.
Kelainan Tulang & Otot Bawaan
KELAINAN WAJAH
Celah Bibir & Celah Langit-langit
Celah Bibir dan Celah Langit-langit adalah suatu kelainan bawaan yang terjadi pada bibir bagian atas serta langit-langit lunak dan langit-langit keras mulut.
Celah bibir (Bibir sumbing) adalah suatu ketidaksempurnaan pada penyambungan bibir bagian atas, yang biasanya berlokasi tepat dibawah hidung.
Celah langit-langit adalah suatu saluran abnormal yang melewati langit-langit mulut dan menuju ke saluran udara di hidung.
Celah bibir dan celah langit-langit bisa terjadi secara bersamaan maupun sendiri-sendiri. Kelainan ini juga bisa terjadi bersamaan dengan kelainan bawaan lainnya.
Penyebabnya mungkin adalah mutasi genetik atau teratogen (zat yang dapat menyebabkan kelainan pada janin, contohnya virus atau bahan kimia).
Selain tidak sedap dipandang, kelainan ini juga menyebabkan anak mengalami kesulitan ketika makan, gangguan perkembangan berbicara dan infeksi telinga.
Faktor resiko untuk kelainan ini adalah riwayat celah bibir atau celah langit-langit pada keluarga serta adanya kelainan bawaan lainnya.

Gejalanya berupa:
- pemisahan bibir
- pemisahan langit-langit
- pemisahan bibir dan langit-langit
- distorsi hidung
- infeksi telinga berulang
- berat badan tidak bertambah
- regurgitasi nasal ketika menyusu (air susu keluar dari lubang hidung).

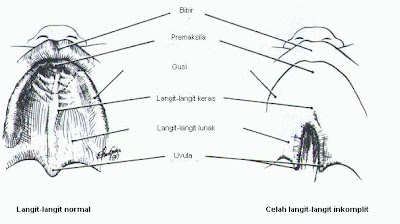
Diagnosis ditegakkan berdasarkan hasil pemeriksaan fisik di daerah wajah.
Pengobatan melibatkan beberapa disiplin ilmu, yaitu bedah plastik, ortodontis, terapi wicara dan lainnya.
Pembedahan untuk menutup celah bibir biasanya dilakukan pada saat anak berusia 3-6 bulan.
Penutupan celah langit-langit biasanya ditunda sampai terjadi perubahan langit-langit yang biasanya berjalan seiring dengan pertumbuhan anak (maksimal sampai anak berumur 1 tahun). Sebelum pembedahan dilakukan, bisa dipasang alat tiruan pada langit-langit mulut untuk membantu pemberian makan/susu.
Pengobatan mungkin berlangsung selama bertahun-tahun dan mungkin perlu dilakukan beberapa kali pembedahan (tergantung kepada luasnya kelainan), tetapi kebanyakan anak akan memiliki penampilan yang normal serta berbicara dan makan secara normal pula. Beberapa diantara mereka mungkin tetap memiliki gangguan berbicara.
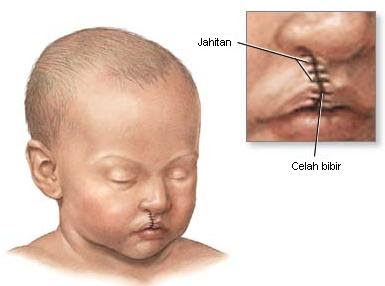
Sindroma Pierre Robin
Sindroma Pierre Robin adalah sekelompok kelainan yang terutama ditandai dengan adanya rahang bawah yang sangat kecil dengan lidah yang jatuh ke belakang dan mengarah ke bawah. Bisa juga disertai dengan tingginya lengkung langit-langit mulut atau celah langit-langit.
Penyebab yang pasti tidak diketahui, bisa merupakan bagian dari sindroma genetik.
Gejalanya berupa:
- rahang yang sangat kecil dengan dagu yang tertarik ke belakang
- lidah tampak besar (sebenarnya ukurannya normal tetapi relatif besar jika dibandingkan dengan rahang yang kecil) dan terletak jauh di belakang orofaring
- lengkung langit-langit yang tinggi
- celah langit-langit lunak
- tercekik/tersedak oleh lidah.
Bayi harus ditempatkan pada posisi membungkuk sehingga gaya tarik bumi akan menarik lidah ke depan dan saluran udara tetap terbuka.
Pada kasus yang agak berat perlu dipasang selang melalui hidung ke saluran udara untuk menghindari penyumbatan saluran udara.
Pada kasus yang berat, jika terjadi penyumbatan saluran udara berulang, perlu dilakukan pembedahan. Kadang perlu dilakukan trakeostomi.
Menyusui atau memberi makan harus dilakukan secara sangat hati-hati untuk menghindari tersedak dan terhirupnya cairan/makanan ke saluran udara,
Tersedak dan gangguan pemberian makan/susu akan berkurang secara spontan, sejalan dengan pertumbuhan rahang.
KELAINAN TULANG BELAKANG
Tortikolis Kongenital
Tortikolis Kongenitalis adalah suatu keadaan dimana leher bayi terpuntir ke salah satu sisi dan kepalanya miring ke sisi tersebut.
Keadaan ini biasanya disebabkan oleh:
# Cedera pada otot atau pembuluh darah leher selama proses persalinan berlangsung
# Kelainan posisi kepala bayi ketika masih berada di dalam rahim
# Sindroma Klippel-Feil (penyatuan tulang belakang leher)
# Fusi atlanto-oksipital (penyatuan tulang belakang leher pertama dengan tulang tengkorak).
Gejalanya bisa berupa:
- pembengkakan otot leher
- kepala miring ke sisi yang terkena
- bahu pada sisi yang terkena tampak terangkat
- otot leher tampak kaku
- pergerakan leher terbatas
- tremor kepala.
Tujuan pengobatan adalah meregangkan otot leher yang memendek.
Pada bayi dan anak kecil dilakukan peregangang pasif. Jika teknik tersebut gagal, pada usia pra-sekolah dilakukan pembedahan.
Skoliosis Kongenitalis
Skoliosis Kongenitalis adalah suatu kelainan pada lengkung tulang belakang bayi baru lahir.
Kelainan ini jarang terjadi dan biasanya berhubungan dengan gangguan pada pembentukan tulang belakang atau peleburan tulang rusuk.
Skoliosis bisa menyebabkan kelainan bentuk yang serius pada anak yang sedang tumbuh, karena itu seringkali dilakukan tindakan pengobatan dengan memasang penyangga (brace) sedini mungkin. Jika keadaan anak semakin memburuk, mungkin perlu dilakukan pembedahan.
KELAINAN PINGGUL, TUNGKAI & KAKI
Dislokasi Pinggul Bawaan
Dislokasi Pinggul Bawaan adalah suatu kelainan bentuk pada persendian pinggul yang ditemukan pada bayi baru lahir atau pada awal masa kanak-kanak.
Pinggul adalah suatu persendian bola dan kantung; bolanya adalah kaput femoralis (kepala tulang paha) yang berada di puncak tulang paha, sedangkan kantungnya adalah asetabulum yang berasal dari panggul.
Penyebabnya tidak diketahui, tetapi diduga melibatkan faktor genetik.
Kelainan yang dirasakan mungkin baru muncul pada usia 30-40 tahun, dan bisa menyerang salah satu maupun kedua pinggul.
Kelainan ini lebih sering ditemui pada:
# Anak pertama
# Bayi perempuan
# Bayi dalam letak bokong
# Riwayat dislokasi pinggul pada keluarga.
Kelainan ini ditemukan pada 1 diantara 1.000 bayi baru lahir.
Gejalanya bisa berupa:
- Pergerakan yang terbatas di daerah yang terkena
- Posisi tungkai yang asimetris
- Lipatan lemak paha yang asimetris
- Setelah bayi berumur 3 bulan : rotasi tungkai asimetris dan tungkai pada sisi yang terkena tampak memendek.
Pemeriksaan yang paling penting adalah USG pinggul. Pada bayi yang lebih besar dan anak-anak bisa dilakukan rontgen pinggul.
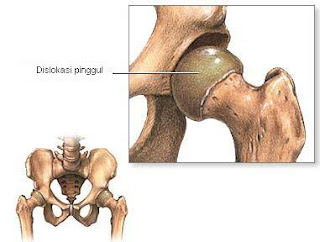
Dislokasi pinggul bawaan
Pada awal masa bayi, agar kaput femoralis tetap berada dalam kantungnya, bisa dipasang alat untuk memisahkan tungkai dan melipatnya ke arah luar (seperti kodok).
Jika posisi diatas sulit dipertahankan, bisa digunakan gips yang secara periodik diganti sehingga pertumbuhan tulang tidak terhambat.
Jika tindakan tersebut tidak berhasil atau jika dislokasi diketahui setelah anak cukup besar, maka dilakukan tindakan pembedahan.
Torsio femoral adalah suatu keadaan dimana lutut menghadap ke depan atau ke samping.
Keadaan ini seringkali membaik dengan sendirinya pada saat anak tumbuh dan mulai berdiri serta berjalan.
Dislokasi lutut adalah suatu keadaan dimana tungkai bawah pada lutut melipat ke depan.
Kelainan ini jarang terjadi tetapi jika terjadi harus segera diatasi. Biasanya dilakukan tindakan menekuk lutut bayi secara perlahan ke depan dan ke belakang sebanyak beberapa kali/hari serta memasang bidai agar lutut tetap tertekuk.
Clubfoot (talipes) adalah suatu keadaan dimana bentuk atau posisi kaki terpuntir.
Lengkung kaki bisa sangat tinggi atau kaki berputar ke dalam maupun ke luar.
Clubfoot sejati disebabkan oleh kelainan anatomis.
Jika tidak terdapat kelainan anatomis, maka keadaan ini bisa diperbaiki dengan pemasangan gips dan terapi fisik. Pengobatan dini dengan gips bisa memperbaiki clubfoot sejati tetapi biasanya perlu dilakukan pembedahan.
AMPUTASI KONGENITAL
Amputasi Kongenital (Missing Limbs) adalah suatu keadaan dimana bayi baru lahir tidak memiliki sebuah lengan atau sebuah tungkai atau bagian dari lengan maupun tungkai.
Penyebabnya tidak diketahui.
Pemakaian talidomid sebagai obat untuk mengatasi morning sickness pada wanita hamil, diduga merupakan penyebab terjadinya kelainan ini.
Agar anggota gerak lebih fungsional, bisa digunakan lengan atau tungkai palsu.
OSTEOGENESIS IMPERFEKTA
Osteogenesis Imperfekta adalah suatu keadaan dimana tulang-tulang menjadi rapuh secara abnormal.
Osteogenesis imperfekta merupakan suatu penyakit keturunan.
Penyakit ini terjadi akibat adanya kelainan pada jumlah atau struktur kolagen tipe I, yang merupakan bagian penting dari tulang.
Osteogenesis imperfekta ditemukan pada 1 diantara 20.000 bayi.
Tulang mudah patah sehingga bayi biasanya terlahir dengan banyak tulang yang patah.
Selama persalinan berlangsung, bisa terjadi trauma kepala dan perdarahan otak karena tulang tengkorak sangat lembut; bayi bisa meninggal dalam beberapa hari setelah lahir.
Banyak bayi yang bertahan hidup, tetapi patah tulang multipel seringkali menyebabkan kelainan bentuk dan dwarfisme (cebol). Jika otaknya tidak terkena, maka kecerdasannya adalah normal.
Trias osteogenesi imperfekta terdiri dari:
- tulang yang rapuh
- gangguan pendengaran
- blue sclerae (bagian putih mata tampak kebiruan).
Tetapi tidak semua penderita memiliki blue sclerae maupun gangguan pendengaran. Semua penderita memiliki tulang yang rapuh, tetapi tidak selalu terjadi patah tulang.
Gejala lainnya yang biasa ditemukan pada osteogenesis imperfekta:
- patah tulang
- pada suatu waktu terjadi lebih dari 1 patah tulang (patah tulang multipel)
- patah tulang bisa terjadi setelah cedera ringan maupun sudah ada ketika bayi lahir
- kelainan bentuk pada anggota gerak
- tuli (gangguan pendengaran konduktif bisa terjadi pada remaja dan dewasa)
- kifosis
- kifoskoliosis
- postur tubuh yang pendek
- kelainan gigi
- pektus karinatum
- pektus ekskavatum (kaki cekung, punggung kaki melengkung sehingga bagian depan punggung kaki menyentuh lantai)
- pes planus (kaki datar, seluruh telapaknya menyentuh lantai)
- persendian yang lemah
- hipermobilitas
- mudah memar
- tungkai melengkung.
Pectus excavatum
Rontgen tulang bisa menunjukkan adanya tanda-tanda patah tulang multipel.
Diagnosis ditegakkan berdasarkan tes kolagen pada biopsi kulit.
Osteogenesis imperfekta yang berat dapat dilihat pada pemeriksaan USG yang dilakukan pada kehamilan 16 minggu.
Untuk mencegah terjadinya kelainan bentuk, setiap patah tulang harus segera diperbaiki.
Gizi yang baik dan latihan yang teratur bisa membantu meningkatkan kekuatan tulang dan otot.
Terapi dan rehabilitasi fisik juga bisa dilakukan. Berenang merupakan olah raga yang baik sekali bagi penderita osteogenesis imperfekta.
Tindakan pembedahan terdiri dari pemasangan batang logam pada tulang agar tulang lebih kuat dan untuk mencegah terjadinya kelainan bentuk.
Beberapa penderita mungkin memerlukan bantuan braces atau tongkat penyangga.
ARTROGIPOSIS MULTIPEL KONGENITAL
Artrogiposis Multipel Kongenital adalah suatu keadaan dimana satu atau beberapa sendi melebur atau mengalami kontraktur (memendek) sehingga tidak dapat ditekuk (pergerakannya terbatas).
Pada beberapa kasus, kelainan ini hanya menyerang sedikit sendi sehingga pergerakan masih mendekati normal.
Pada kasus yang klasik, kelainan ini ditemukan pada tangan, pergelangan tangan, sikut, bahu, pinggul, kaki dan lutut.
Pada kasus yang sangat berat, hampir semua sendi terkena, termasuk sendi rahang dan punggung.
Kontraktur sendi seringkali disertai dengan kelemahan otot sehingga pergerakan penderita menjadi lebih terbatas.
Artrogriposis ditemukan pada 1 diantara 3.000 bayi baru lahir dan bukan merupakan penyakit keturunan.
Secara umum, terdapat 4 penyebab dari terbatasnya pergerakan sendi bawaan:
1. Atrofi otot (pengkisutan otot atau otot tidak terbentuk sebagaimana mestinya). Penyebab yang pasti dari terjadinya atrofi otot tidak diketahui, tetapi diduga disebabkan oleh:
- Penyakit otot (misalnya distrofi muskuler kongenital)
- Demam pada ibu hamil
- Infeksi virus selama hamil, yang bisa menyebabkan kerusakan sel yang menghantarkan gelombang saraf ke otot.
2. Rahim terlalu sempit untuk pergerakan yang normal. Misalnya jumlah cairan ketuban yang kurang atau bentuk rahim yang tidak normal.
3. Kelainan bentuk pada sistem saraf pusat dan korda spinalis. Pada kasus ini, biasanya artrogiposis disertai oleh kelainan lainnya.
4. Gangguan pembentukan tendo, tulang, sendi atau lapisan sendi. Misalnya tendo tidak tersambung dengan sendi pada tempat yang semestinya.
Untuk memperbaiki kekuatan otot dan pergerakan sendi, dilakukan terapi fisik.
Pemasangan bidai bisa dilakukan untuk meningkatkan latihan peregangan sehingga sendi lebih mudah digerakkan.
Untuk memperbaiki posisi kaki seringkali digunakan gips.
Pembedahan sebaiknya digunakan sebagai tindakan suportif terhadap tindakan pengobatan lainnya setelah dicapai hasil yang maksimum.
Pembedahan biasanya dilakukan pada pergelangan kaki untuk mengembalikan posisi kaki sehingga bisa menahan beban dan berjalan. Kadang pembedahan dilakukan pada lutut, pinggul, sikut dan pergelangan tangan agar posisinya lebih baik atau gerakannya lebih luas.
Pada beberapa kasus dilakukan pemindahan tendo untuk memperbaiki fungsi otot.
Artrogriposis merupakan suatu kelainan yang non-progresif, artinya tidak semakin memburuk sejalan dengan bertambahnya usia anak. Bahkan dengan terapi fisik dan pengobatan lainnya, kemungkinan akan terjadi perbaikan fungsi yang berarti.
Kebanyakan penderita memiliki tingkat kecerdasan yang normal dan ketika dewasa mampu hidup produktif dan mandiri,
KELAINAN OTOT
Otot pektoralis mayor di dada bisa tidak terbentuk sama sekali atau hanya terbentuk sebagian.
Kelainan ini bisa berdiri sendiri atau terjadi bersamaan dengan kelainan pada tangan.
Sindroma Prune-belly
Sindroma Prune-belly (Sindroma Eagle-Barrett) adalah suatu penyakit yang ditandai dengan trias:
# Kelainan otot perut (menyebabkan kulit perut mengkerut seperti buah plum yang dikeringkan)
# Undesensus testis (buah zakar tidak turun ke dalam kantung zakar)
# Pelebaran saluran kemih.
Penyebabnya tidak diketahui.
Selama masa janin, terdapat penyumbatan pada aliran air kemih dari ginjal.
Kelainan ini ditemukan pada 1 dari 30.000-40.000 bayi baru lahir. Lebih banyak ditemukan pada bayi laki-laki.
Ibu yang mengandung bayi dengan kelainan ini bisa mengalami oligohidramnion (kekurangan cairan ketuban) yang merupakan faktor predisposisi untuk terjadinya gangguan paru-paru pada bayi.
Perut bayi tampak mengkerut, hal ini diperkuat dengan adanya kekurangan otot perut.
Kelainan alat kelamin tampak sebagai undesensus testis.
Hampir sepertiga bayi meninggal ketika lahir atau beberapa minggu kemudian akibat kelainan paru-paru, ginjal maupun kelainan kedua organ tersebut yang sifatnya berat.
Kelainan lainnya yang mungkin ditemukan adalah kelainan muskuloskeletal (otot dan kerangka tubuh), kelainan jantung dan kelainan usus.
Gejalanya berupa:
- oligohidramnion (air ketuban sedikit atau tidak ada)
- club foot.
Kelainan sistem ginjal yang ditemukan pada pemeriksaan fisik maupun pemeriksaan laboratorium:
- pelebaran struktur pengumpul pada ginjal
- megaureter (pelebaran ureter)
- pelebaran kandung kemih
- megalouretra (pelebaran uretra).
Pemeriksaan yang biasa dilakukan:
- USG menunjukkan pelebaran ginjal, ureter atau kandung kemih
- Rontgen ginjal
- IVP (pielogram intravena)
- Rontgen dada.
Untuk mempertahankan pengaliran sistem ginjal dan melindungi fungsi yang masih tersisa, dilakukan pembedahan, yang bisa berupa:
# Pielostomi (membuat lubang ke dalam ginjal, biasanya disertai dengan pemasangan selang drainase)
# Vesikostomi (membuat lubang ke dalam kandung kemih, biasanya disertai dengan pemasangan selang drainase)
# Bedah rekonstruktif
# Bedah prenatal untuk memperbaiki katup uretra posterior yang tidak membuka.
Pengobatan lainnya:
- Antibiotik prefentif untuk mencegah infeksi saluran kemih
- Antibiotik untuk infeksi akut
- Orkidopeksi untuk undesensus testis.
Kelainan Tulang & Otot Bawaan
KELAINAN WAJAH
Celah Bibir & Celah Langit-langit
Celah Bibir dan Celah Langit-langit adalah suatu kelainan bawaan yang terjadi pada bibir bagian atas serta langit-langit lunak dan langit-langit keras mulut.
Celah bibir (Bibir sumbing) adalah suatu ketidaksempurnaan pada penyambungan bibir bagian atas, yang biasanya berlokasi tepat dibawah hidung.
Celah langit-langit adalah suatu saluran abnormal yang melewati langit-langit mulut dan menuju ke saluran udara di hidung.
Celah bibir dan celah langit-langit bisa terjadi secara bersamaan maupun sendiri-sendiri. Kelainan ini juga bisa terjadi bersamaan dengan kelainan bawaan lainnya.
Penyebabnya mungkin adalah mutasi genetik atau teratogen (zat yang dapat menyebabkan kelainan pada janin, contohnya virus atau bahan kimia).
Selain tidak sedap dipandang, kelainan ini juga menyebabkan anak mengalami kesulitan ketika makan, gangguan perkembangan berbicara dan infeksi telinga.
Faktor resiko untuk kelainan ini adalah riwayat celah bibir atau celah langit-langit pada keluarga serta adanya kelainan bawaan lainnya.
Celah bibir
Gejalanya berupa:
- pemisahan bibir
- pemisahan langit-langit
- pemisahan bibir dan langit-langit
- distorsi hidung
- infeksi telinga berulang
- berat badan tidak bertambah
- regurgitasi nasal ketika menyusu (air susu keluar dari lubang hidung).
Diagnosis ditegakkan berdasarkan hasil pemeriksaan fisik di daerah wajah.
Pengobatan melibatkan beberapa disiplin ilmu, yaitu bedah plastik, ortodontis, terapi wicara dan lainnya.
Pembedahan untuk menutup celah bibir biasanya dilakukan pada saat anak berusia 3-6 bulan.
Penutupan celah langit-langit biasanya ditunda sampai terjadi perubahan langit-langit yang biasanya berjalan seiring dengan pertumbuhan anak (maksimal sampai anak berumur 1 tahun). Sebelum pembedahan dilakukan, bisa dipasang alat tiruan pada langit-langit mulut untuk membantu pemberian makan/susu.
Pengobatan mungkin berlangsung selama bertahun-tahun dan mungkin perlu dilakukan beberapa kali pembedahan (tergantung kepada luasnya kelainan), tetapi kebanyakan anak akan memiliki penampilan yang normal serta berbicara dan makan secara normal pula. Beberapa diantara mereka mungkin tetap memiliki gangguan berbicara.
Sindroma Pierre Robin
Sindroma Pierre Robin adalah sekelompok kelainan yang terutama ditandai dengan adanya rahang bawah yang sangat kecil dengan lidah yang jatuh ke belakang dan mengarah ke bawah. Bisa juga disertai dengan tingginya lengkung langit-langit mulut atau celah langit-langit.
Penyebab yang pasti tidak diketahui, bisa merupakan bagian dari sindroma genetik.
Gejalanya berupa:
- rahang yang sangat kecil dengan dagu yang tertarik ke belakang
- lidah tampak besar (sebenarnya ukurannya normal tetapi relatif besar jika dibandingkan dengan rahang yang kecil) dan terletak jauh di belakang orofaring
- lengkung langit-langit yang tinggi
- celah langit-langit lunak
- tercekik/tersedak oleh lidah.
Bayi harus ditempatkan pada posisi membungkuk sehingga gaya tarik bumi akan menarik lidah ke depan dan saluran udara tetap terbuka.
Pada kasus yang agak berat perlu dipasang selang melalui hidung ke saluran udara untuk menghindari penyumbatan saluran udara.
Pada kasus yang berat, jika terjadi penyumbatan saluran udara berulang, perlu dilakukan pembedahan. Kadang perlu dilakukan trakeostomi.
Menyusui atau memberi makan harus dilakukan secara sangat hati-hati untuk menghindari tersedak dan terhirupnya cairan/makanan ke saluran udara,
Tersedak dan gangguan pemberian makan/susu akan berkurang secara spontan, sejalan dengan pertumbuhan rahang.
KELAINAN TULANG BELAKANG
Tortikolis Kongenital
Tortikolis Kongenitalis adalah suatu keadaan dimana leher bayi terpuntir ke salah satu sisi dan kepalanya miring ke sisi tersebut.
Keadaan ini biasanya disebabkan oleh:
# Cedera pada otot atau pembuluh darah leher selama proses persalinan berlangsung
# Kelainan posisi kepala bayi ketika masih berada di dalam rahim
# Sindroma Klippel-Feil (penyatuan tulang belakang leher)
# Fusi atlanto-oksipital (penyatuan tulang belakang leher pertama dengan tulang tengkorak).
Gejalanya bisa berupa:
- pembengkakan otot leher
- kepala miring ke sisi yang terkena
- bahu pada sisi yang terkena tampak terangkat
- otot leher tampak kaku
- pergerakan leher terbatas
- tremor kepala.
Tujuan pengobatan adalah meregangkan otot leher yang memendek.
Pada bayi dan anak kecil dilakukan peregangang pasif. Jika teknik tersebut gagal, pada usia pra-sekolah dilakukan pembedahan.
Skoliosis Kongenitalis
Skoliosis Kongenitalis adalah suatu kelainan pada lengkung tulang belakang bayi baru lahir.
Kelainan ini jarang terjadi dan biasanya berhubungan dengan gangguan pada pembentukan tulang belakang atau peleburan tulang rusuk.
Skoliosis bisa menyebabkan kelainan bentuk yang serius pada anak yang sedang tumbuh, karena itu seringkali dilakukan tindakan pengobatan dengan memasang penyangga (brace) sedini mungkin. Jika keadaan anak semakin memburuk, mungkin perlu dilakukan pembedahan.
KELAINAN PINGGUL, TUNGKAI & KAKI
Dislokasi Pinggul Bawaan
Dislokasi Pinggul Bawaan adalah suatu kelainan bentuk pada persendian pinggul yang ditemukan pada bayi baru lahir atau pada awal masa kanak-kanak.
Pinggul adalah suatu persendian bola dan kantung; bolanya adalah kaput femoralis (kepala tulang paha) yang berada di puncak tulang paha, sedangkan kantungnya adalah asetabulum yang berasal dari panggul.
Penyebabnya tidak diketahui, tetapi diduga melibatkan faktor genetik.
Kelainan yang dirasakan mungkin baru muncul pada usia 30-40 tahun, dan bisa menyerang salah satu maupun kedua pinggul.
Kelainan ini lebih sering ditemui pada:
# Anak pertama
# Bayi perempuan
# Bayi dalam letak bokong
# Riwayat dislokasi pinggul pada keluarga.
Kelainan ini ditemukan pada 1 diantara 1.000 bayi baru lahir.
Gejalanya bisa berupa:
- Pergerakan yang terbatas di daerah yang terkena
- Posisi tungkai yang asimetris
- Lipatan lemak paha yang asimetris
- Setelah bayi berumur 3 bulan : rotasi tungkai asimetris dan tungkai pada sisi yang terkena tampak memendek.
Pemeriksaan yang paling penting adalah USG pinggul. Pada bayi yang lebih besar dan anak-anak bisa dilakukan rontgen pinggul.
Dislokasi pinggul bawaan
Pada awal masa bayi, agar kaput femoralis tetap berada dalam kantungnya, bisa dipasang alat untuk memisahkan tungkai dan melipatnya ke arah luar (seperti kodok).
Jika posisi diatas sulit dipertahankan, bisa digunakan gips yang secara periodik diganti sehingga pertumbuhan tulang tidak terhambat.
Jika tindakan tersebut tidak berhasil atau jika dislokasi diketahui setelah anak cukup besar, maka dilakukan tindakan pembedahan.
Torsio femoral adalah suatu keadaan dimana lutut menghadap ke depan atau ke samping.
Keadaan ini seringkali membaik dengan sendirinya pada saat anak tumbuh dan mulai berdiri serta berjalan.
Dislokasi lutut adalah suatu keadaan dimana tungkai bawah pada lutut melipat ke depan.
Kelainan ini jarang terjadi tetapi jika terjadi harus segera diatasi. Biasanya dilakukan tindakan menekuk lutut bayi secara perlahan ke depan dan ke belakang sebanyak beberapa kali/hari serta memasang bidai agar lutut tetap tertekuk.
Clubfoot (talipes) adalah suatu keadaan dimana bentuk atau posisi kaki terpuntir.
Lengkung kaki bisa sangat tinggi atau kaki berputar ke dalam maupun ke luar.
Clubfoot sejati disebabkan oleh kelainan anatomis.
Jika tidak terdapat kelainan anatomis, maka keadaan ini bisa diperbaiki dengan pemasangan gips dan terapi fisik. Pengobatan dini dengan gips bisa memperbaiki clubfoot sejati tetapi biasanya perlu dilakukan pembedahan.
AMPUTASI KONGENITAL
Amputasi Kongenital (Missing Limbs) adalah suatu keadaan dimana bayi baru lahir tidak memiliki sebuah lengan atau sebuah tungkai atau bagian dari lengan maupun tungkai.
Penyebabnya tidak diketahui.
Pemakaian talidomid sebagai obat untuk mengatasi morning sickness pada wanita hamil, diduga merupakan penyebab terjadinya kelainan ini.
Agar anggota gerak lebih fungsional, bisa digunakan lengan atau tungkai palsu.
OSTEOGENESIS IMPERFEKTA
Osteogenesis Imperfekta adalah suatu keadaan dimana tulang-tulang menjadi rapuh secara abnormal.
Osteogenesis imperfekta merupakan suatu penyakit keturunan.
Penyakit ini terjadi akibat adanya kelainan pada jumlah atau struktur kolagen tipe I, yang merupakan bagian penting dari tulang.
Osteogenesis imperfekta ditemukan pada 1 diantara 20.000 bayi.
Tulang mudah patah sehingga bayi biasanya terlahir dengan banyak tulang yang patah.
Selama persalinan berlangsung, bisa terjadi trauma kepala dan perdarahan otak karena tulang tengkorak sangat lembut; bayi bisa meninggal dalam beberapa hari setelah lahir.
Banyak bayi yang bertahan hidup, tetapi patah tulang multipel seringkali menyebabkan kelainan bentuk dan dwarfisme (cebol). Jika otaknya tidak terkena, maka kecerdasannya adalah normal.
Trias osteogenesi imperfekta terdiri dari:
- tulang yang rapuh
- gangguan pendengaran
- blue sclerae (bagian putih mata tampak kebiruan).
Tetapi tidak semua penderita memiliki blue sclerae maupun gangguan pendengaran. Semua penderita memiliki tulang yang rapuh, tetapi tidak selalu terjadi patah tulang.
Gejala lainnya yang biasa ditemukan pada osteogenesis imperfekta:
- patah tulang
- pada suatu waktu terjadi lebih dari 1 patah tulang (patah tulang multipel)
- patah tulang bisa terjadi setelah cedera ringan maupun sudah ada ketika bayi lahir
- kelainan bentuk pada anggota gerak
- tuli (gangguan pendengaran konduktif bisa terjadi pada remaja dan dewasa)
- kifosis
- kifoskoliosis
- postur tubuh yang pendek
- kelainan gigi
- pektus karinatum
- pektus ekskavatum (kaki cekung, punggung kaki melengkung sehingga bagian depan punggung kaki menyentuh lantai)
- pes planus (kaki datar, seluruh telapaknya menyentuh lantai)
- persendian yang lemah
- hipermobilitas
- mudah memar
- tungkai melengkung.
Pectus excavatum
Rontgen tulang bisa menunjukkan adanya tanda-tanda patah tulang multipel.
Diagnosis ditegakkan berdasarkan tes kolagen pada biopsi kulit.
Osteogenesis imperfekta yang berat dapat dilihat pada pemeriksaan USG yang dilakukan pada kehamilan 16 minggu.
Untuk mencegah terjadinya kelainan bentuk, setiap patah tulang harus segera diperbaiki.
Gizi yang baik dan latihan yang teratur bisa membantu meningkatkan kekuatan tulang dan otot.
Terapi dan rehabilitasi fisik juga bisa dilakukan. Berenang merupakan olah raga yang baik sekali bagi penderita osteogenesis imperfekta.
Tindakan pembedahan terdiri dari pemasangan batang logam pada tulang agar tulang lebih kuat dan untuk mencegah terjadinya kelainan bentuk.
Beberapa penderita mungkin memerlukan bantuan braces atau tongkat penyangga.
ARTROGIPOSIS MULTIPEL KONGENITAL
Artrogiposis Multipel Kongenital adalah suatu keadaan dimana satu atau beberapa sendi melebur atau mengalami kontraktur (memendek) sehingga tidak dapat ditekuk (pergerakannya terbatas).
Pada beberapa kasus, kelainan ini hanya menyerang sedikit sendi sehingga pergerakan masih mendekati normal.
Pada kasus yang klasik, kelainan ini ditemukan pada tangan, pergelangan tangan, sikut, bahu, pinggul, kaki dan lutut.
Pada kasus yang sangat berat, hampir semua sendi terkena, termasuk sendi rahang dan punggung.
Kontraktur sendi seringkali disertai dengan kelemahan otot sehingga pergerakan penderita menjadi lebih terbatas.
Artrogriposis ditemukan pada 1 diantara 3.000 bayi baru lahir dan bukan merupakan penyakit keturunan.
Secara umum, terdapat 4 penyebab dari terbatasnya pergerakan sendi bawaan:
1. Atrofi otot (pengkisutan otot atau otot tidak terbentuk sebagaimana mestinya). Penyebab yang pasti dari terjadinya atrofi otot tidak diketahui, tetapi diduga disebabkan oleh:
- Penyakit otot (misalnya distrofi muskuler kongenital)
- Demam pada ibu hamil
- Infeksi virus selama hamil, yang bisa menyebabkan kerusakan sel yang menghantarkan gelombang saraf ke otot.
2. Rahim terlalu sempit untuk pergerakan yang normal. Misalnya jumlah cairan ketuban yang kurang atau bentuk rahim yang tidak normal.
3. Kelainan bentuk pada sistem saraf pusat dan korda spinalis. Pada kasus ini, biasanya artrogiposis disertai oleh kelainan lainnya.
4. Gangguan pembentukan tendo, tulang, sendi atau lapisan sendi. Misalnya tendo tidak tersambung dengan sendi pada tempat yang semestinya.
Untuk memperbaiki kekuatan otot dan pergerakan sendi, dilakukan terapi fisik.
Pemasangan bidai bisa dilakukan untuk meningkatkan latihan peregangan sehingga sendi lebih mudah digerakkan.
Untuk memperbaiki posisi kaki seringkali digunakan gips.
Pembedahan sebaiknya digunakan sebagai tindakan suportif terhadap tindakan pengobatan lainnya setelah dicapai hasil yang maksimum.
Pembedahan biasanya dilakukan pada pergelangan kaki untuk mengembalikan posisi kaki sehingga bisa menahan beban dan berjalan. Kadang pembedahan dilakukan pada lutut, pinggul, sikut dan pergelangan tangan agar posisinya lebih baik atau gerakannya lebih luas.
Pada beberapa kasus dilakukan pemindahan tendo untuk memperbaiki fungsi otot.
Artrogriposis merupakan suatu kelainan yang non-progresif, artinya tidak semakin memburuk sejalan dengan bertambahnya usia anak. Bahkan dengan terapi fisik dan pengobatan lainnya, kemungkinan akan terjadi perbaikan fungsi yang berarti.
Kebanyakan penderita memiliki tingkat kecerdasan yang normal dan ketika dewasa mampu hidup produktif dan mandiri,
KELAINAN OTOT
Otot pektoralis mayor di dada bisa tidak terbentuk sama sekali atau hanya terbentuk sebagian.
Kelainan ini bisa berdiri sendiri atau terjadi bersamaan dengan kelainan pada tangan.
Sindroma Prune-belly
Sindroma Prune-belly (Sindroma Eagle-Barrett) adalah suatu penyakit yang ditandai dengan trias:
# Kelainan otot perut (menyebabkan kulit perut mengkerut seperti buah plum yang dikeringkan)
# Undesensus testis (buah zakar tidak turun ke dalam kantung zakar)
# Pelebaran saluran kemih.
Penyebabnya tidak diketahui.
Selama masa janin, terdapat penyumbatan pada aliran air kemih dari ginjal.
Kelainan ini ditemukan pada 1 dari 30.000-40.000 bayi baru lahir. Lebih banyak ditemukan pada bayi laki-laki.
Ibu yang mengandung bayi dengan kelainan ini bisa mengalami oligohidramnion (kekurangan cairan ketuban) yang merupakan faktor predisposisi untuk terjadinya gangguan paru-paru pada bayi.
Perut bayi tampak mengkerut, hal ini diperkuat dengan adanya kekurangan otot perut.
Kelainan alat kelamin tampak sebagai undesensus testis.
Hampir sepertiga bayi meninggal ketika lahir atau beberapa minggu kemudian akibat kelainan paru-paru, ginjal maupun kelainan kedua organ tersebut yang sifatnya berat.
Kelainan lainnya yang mungkin ditemukan adalah kelainan muskuloskeletal (otot dan kerangka tubuh), kelainan jantung dan kelainan usus.
Gejalanya berupa:
- oligohidramnion (air ketuban sedikit atau tidak ada)
- club foot.
Kelainan sistem ginjal yang ditemukan pada pemeriksaan fisik maupun pemeriksaan laboratorium:
- pelebaran struktur pengumpul pada ginjal
- megaureter (pelebaran ureter)
- pelebaran kandung kemih
- megalouretra (pelebaran uretra).
Pemeriksaan yang biasa dilakukan:
- USG menunjukkan pelebaran ginjal, ureter atau kandung kemih
- Rontgen ginjal
- IVP (pielogram intravena)
- Rontgen dada.
Untuk mempertahankan pengaliran sistem ginjal dan melindungi fungsi yang masih tersisa, dilakukan pembedahan, yang bisa berupa:
# Pielostomi (membuat lubang ke dalam ginjal, biasanya disertai dengan pemasangan selang drainase)
# Vesikostomi (membuat lubang ke dalam kandung kemih, biasanya disertai dengan pemasangan selang drainase)
# Bedah rekonstruktif
# Bedah prenatal untuk memperbaiki katup uretra posterior yang tidak membuka.
Pengobatan lainnya:
- Antibiotik prefentif untuk mencegah infeksi saluran kemih
- Antibiotik untuk infeksi akut
- Orkidopeksi untuk undesensus testis.
Defek Septum Atrium (ASD, Atrial Septal Defect)

PENYEBAB
ASD merupakan suatu kelainan jantung bawaan.
Dalam keadaan normal, pada peredaran darah janin terdapat suatu lubang diantara atrium kiri dan kanan sehingga darah tidak perlu melewati paru-paru. Pada saat bayi lahir, lubang ini biasanya menutup. Jika lubang ini tetap terbuka, darah terus mengalir dari atrium kiri ke atrium kanan (shunt).
Penyebab dari tidak menutupnya lubang pada septum atrium ini tidak diketahui.
GEJALA
Gejalanya bisa berupa:
-sering mengalami infeksi saluran pernafasan
- dispneu (kesulitan dalam bernafas)
- sesak nafas ketika melakukan aktivitas
- jantung berdebar-debar (palpitasi).
Pada kelainan yang sifatnya ringan sampai sedang, mungkin sama sekali tidak ditemukan gejala atau gejalanya baru timbul pada usia pertengahan.
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik:
- denyut arteri pulmonalis dapat diraba di dada
- pemeriksaan dengan stetoskop menunjukkan bunyi jantung yang abnormal. Bisa terdengar murmur akibat peningkatan aliran darah yang malalui katup pulmonalis
- tanda-tanda gagal jantung.
- jika shuntnya besar, murmur juga bisa terdengar akibat peningkatan aliran darah yang mengalir melalui katup trikuspidalis.
Pemeriksaan yang biasa dilakukan:
# Rontgen dada
# Ekokardiografi
# Doppler berwarna
# Ekokardiografi transesofageal
# Kateterisasi jantung
# Angiografi koroner (untuk penderita diatas 35 tahun)
# MRI dada
# EKG menunjukkan adanya fibrilasi atrium atau pembesaran atrium kanan.
PENGOBATAN
Menutup ASD pada masa kanak-kanak bisa mencegah terjadinya kelainan yang serius di kemudian hari.
Jika gejalanya ringan atau tidak ada gejala, tidak perlu dilakukan pengobatan.
Jika lubangnya besar atau terdapat gejala, dilakukan pembedahan untuk menutup ASD.
Pengobatan pencegahan dengan antibiotik sebaiknya diberikan setiap kali sebelum penderita menjalani tindakan pencabutan gigi untuk mengurangi resiko terjadinya endokarditis infektif.
Kelainan Otak Bawaan
Kelainan otak bisa terjadi pada saat otak sedang dibentuk maupun setelah otak terbentuk sempurna.
Beberapa kelainan otak dapat diketahui sebelum bayi lahir, yaitu melalui pemeriksaan USG dan pemeriksaan cairan ketuban.
ANENSEFALUS
Anensefalus adalah suatu keadaan dimana sebagian besar tulang tengkorak dan otak tidak terbentuk.
Anensefalus adalah suatu kelainan tabung saraf (suatu kelainan yang terjadi pada awal perkembangan janin yang menyebabkan kerusakan pada jaringan pembentuk otak dan korda spinalis).
Anensefalus terjadi jika tabung saraf sebelah atas gagal menutup, tetapi penyebabnya yang pasti tidak diketahui.
Penelitian menunjukkan kemungkinan anensefalus berhubungan dengan racun di lingkungan juga kadar asam folat yang rendah dalam darah.
Anensefalus ditemukan pada 3,6-4,6 dari 10.000 bayi baru lahir.
Faktor resiko terjadinya anensefalus adalah:
- Riwayat anensefalus pada kehamilan sebelumnya
- Kadar asam folat yang rendah.
Resiko terjadinya anensefalus bisa dikurangi dengan cara meningkatkan asupan asam folat minimal 3 bulan sebelum hamil dan selama kehamilan bulan pertama.
Gejalanya berupa:
# Ibu : polihidramnion (cairan ketuban di dalam rahim terlalu banyak)
# Bayi
- tidak memiliki tulang tengkorak
- tidak memiliki otak (hemisfer serebri dan serebelum)
- kelainan pada gambaran wajah
- kelainan jantung.
Pemeriksaan yang biasa dilakukan adalah:
- Kadar asam lemak dalam serum ibu hamil
- Amniosentesis (untuk mengetahui adanya peningkatan kadar alfa-fetoprotein)
- Kadar alfa-fetoprotein meningkat (menunjukkan adanya kelainan tabung saraf)
- Kadar estriol pada air kemih ibu
- USG.
Bayi yang menderita anensefalus tidak akan bertahan, mereka lahir dalam keadaan meninggal atau akan meninggal dalam waktu beberapa hari setelah lahir.
MIKROSEFALUS
Mikrosefalus adalah suatu keadaan dimana ukuran kepala (lingkar puncak kepala) lebih kecil dari ukuran kepala rata-rata pada bayi berdasarkan umur dan jenis kelamin.
Dikatakan lebih kecil jika ukuran lingkar kepala kurang dari 42 cm atau lebih kecil dari standar deviasi 3 dibawah angka rata-rata.
Mikrosefalus seringkali terjadi akibat kegagalan pertumbuhan otak pada kecepatan yang normal. Berbagai keadaan dan penyakit yang mempengaruhi pertumbuhan otak bisa menyebabkan mikrosefalus.
Mikrosefalus seringkali berhubungan dengan keterbelakangan mental.
Mikrosefalus bisa terjadi setelah infeksi yang menyebabkan kerusakan pada otak pada bayi yang sangat muda (misalnya meningitis dan meningoensefalitis).
Penyebab utama:
# Sindroma Down
# Sindroma cri du chat
# Sindroma Seckel
# Sindroma Rubinstein-Taybi
# Trisomi 13
# Trisomi 18
# Sindroma Smith-Lemli-Opitz
# Sindroma Cornelia de Lange
Penyebab sekunder:
# Fenilketonuria pada ibu yang tidak terkontrol
# Keracunan metil merkuri
# Rubella kongenital
# Toksoplasmosis kongenital
# Sitomegalovirus kongenital
# Penyalahgunaan obat oleh ibu hamil
# Kekurangan gizi (malnutrisi).
Perawatan pada mikrosefalus tergantung kepada penyebabnya.
Bayi yang menderita mikrosefalus seringkali bisa bertahan hidup tetapi cenderung mengalami keterbelakangan mental, gangguan koordinasi otot dan kejang.
ENSEFALOKEL
Ensefalokel adalah suatu kelainan tabung saraf yang ditandai dengan adanya penonjolan meningens (selaput otak) dan otak yang berbentuk seperti kantung melalui suatu lubang pada tulang tengkorak.
Ensefalokel disebabkan oleh kegagalan penutupan tabung saraf selama perkembangan janin.
Gejalanya berupa:
- hidrosefalus
- kelumpuhan keempat anggota gerak (kuadriplegia spastik)
- gangguan perkembangan
- mikrosefalus
- gangguan penglihatan
- keterbelakangan mental dan pertumbuhan
- ataksia
- kejang.
Beberapa anak memiliki kecerdasan yang normal.
Ensefalokel seringkali disertai dengan kelainan kraniofasial atau kelainan otak lainnya.
Biasanya dilakukan pembedahan untuk mengembalikan jaringan otak yang menonjol ke dalam tulang tengkorak, membuang kantung dan memperbaiki kelainan kraniofasial yang terjadi.
Untuk hidrosefalus mungkin perlu dibuat suatu shunt. Pengobatan lainnya bersifat simtomatis dan suportif.
Prognosisnya tergantung kepada jaringan otak yang terkena, lokasi kantung dan kelainan otak yang menyertainya.
PORENSEFALUS
Porensefalus adalah suatu keadaan dimana pada hemisfer serebri ditemukan suatu kista atau rongga abnormal.
Porensefalus merupakan akibat dari kerusakan otak dan biasanya berhubungan dengan kelainan fungsi otak. Tetapi beberapa anak yang menderita porensefalus memiliki kecerdasan yang normal.
HIDRANENSEFALUS
Hidranensefalus adalah suatu keadaan dimana hemisfer serebri tidak ada dan digantikan oleh kantung-kantung yang berisi cairan serebrospinalis.
Ketika lahir, bayi tampak normal. Ukuran kepala dan refleks spontannya (misalnya refleks menghisap, menelan, menangis dan menggerakkan lengan dan tungkainya) tampak normal. Tetapi beberapa minggu kemudian, biasanya bayi menjadi rewel dan ketegangan ototnya meningkat.
Setelah berusia beberapa bulan, bisa terjadi kejang dan hidrosefalus. Gejala lainnya adalah:
- gangguan penglihatan
- gangguan pertumbuhan
- tuli
- buta
- kelumpuhan (kuadriplegi spastis)
- gangguan kecerdasan.
Hidranensefalus merupakan bentuk porensefalus yang ekstrim dan bisa disebabkan oleh infeksi pembuluh darah atau cedera yang terjadi pada saat usia kehamilan telah mencapai 12 minggu.
Diagnosisnya mungkin tertunda selama beberapa bulan karena perilaku awalnya relatif normal. Beberapa bayi menunjukkan kelainan pada saat lahir, yaitu berupa kejang, mioklonus (kejang atau kedutan otot atau sekelompok otot) dan gangguan pernafasan.
Tidak ada pengobatan yang pasti untuk hidranensefalus. Pengobatannya bersifat simtomatis dan suportif. Prognosis hidranensefalus adalah jelek, bayi biasanya meninggal sebelum berumur 1 tahun.
Hidrosefalus bisa diatasi dengan membuat shunt.
HIDROSEFALUS
Hidrosefalus adalah penimbunan cairan serebrospinal yang berlebihan di dalam otak.
Cairan serebrospinal dibuat di dalam otak dan biasanya beredar ke seluruh bagian otak, selaput otak serta kanalis spinalis, kemudian diserap ke dalam sistem peredaran darah.
Jika terjadi gangguan pada peredaran maupun penyerapan cairan serebrospinal, atau jika cairan yang dibentuk terlalu banyak, maka volume cairan di dalam otak menjadi lebih tinggi dari normal. Penimbunan cairan menyebabkan penekanan pada otak sehingga memaksa otak untuk mendorong tulang tengkorak atau merusak jaringan otak.
Gejalanya bervariasi, tergantung kepada penyebab dari penyumbatan aliran cairan serebrospinal dan luasnya kerusakan jaringan otak akibat hidrosefalus.
Pada bayi, cairan menumpuk pada sistem saraf pusat dan menyebabkan ubun-ubun menonjol serta kepala membesar. Kepala bisa membesar karena piringan tulang tengkorak belum sepenuhnya menutup. Tetapi, jika tulang tengkorak telah menutup (sekitar usia 5 tahun), maka tulang tengkorak tidak dapat membesar lagi.
Pada anak-anak, resiko terjadinya hidrosefalus ditemukan pada:
# Kelainan bawaan
# Tumor pada sistem saraf pusat
# Infeksi dalam kandungan
# Infeksi sistem saraf pusat pada bayi atau anak-anak (misalnya meningitis atau ensefalitis)
# Cedera pada proses kelahiran
# Cedera sebelum atau sesudah lahir (misalnya perdarahan subaraknoid).
Mielomeningokel adalah suatu penyakit dimana terjadi penutupan yan g tidak sempurna pada kolumna spinalis dan berhubungan erat dengan hidrosefalus.
Pada anak-anak yang lebih besar, resiko terjadinya hidrosefalus adalah:
# Riwayat kelainan bawaan
# Space-occupying lesions atau tumor otak maupun korda spinalis
# Infeksi sistem saraf pusat
# Perdarahan otak
# Trauma.
Gejala awal pada bayi:
- Kepala membesar
- Ubun-ubun menonjol dengan atau tanpa pembesaran kepala
- Sutura terpisah.
Gejala pada hidrosefalus lanjutan:
- Rewel, tidak dapat menahan emosi
- Kejang otot.
Gejala lanjut:
- Penurunan fungsi mental
- Gangguan perkembangan
- Penurunan pergerakan
- Gerakan menjadi lambat atau terhambat
- Tidak mau makan/menyusu
- Lemas, tidur terus
- Beser
- Menangis dengan nada tinggi, keras dan singkat
- Gangguan pertumbuhan.
Gejala pada bayi yang lebih tua dan anak-anak bervariasi, tergantung kepada luas kerusakan akibat penekanan terhadap otak. Gejalanya mirip dengan gejala hidrosefalus lanjutan pada anak-anak atau bisa berupa:
- Sakit kepala
- Muntah
- Gangguan penglihatan
- Juling
- Pergerakan mata yang tidak terkendali
- Hilangnya koordinasi
- Langkahnya tidak tegas
- Kelainan mental (misalnya psikosa).
Mengetuk tulang tengkorak secara perlahan dengan ujung jari tangan akan menunjukkan suara abnormal akibat penipisan dan pemisahan tulang-tulang tengkorak.
Vena pada tulang tengkorak tampak melebar. Lingkar kepala membesar atau kepala membesar hanya pada bagian tertentu (paling sering di daerah dahi). Mata tampak tertekan (gambaran sunset, dimana bagian putih mata terlihat diatas iris).
Pemeriksaan saraf menunjukkan adanya defisit neurologis fokal (gangguan fungsi saraf yang sifatnya lokal) dan refleks yang abnormal.
Pemeriksaan yang biasa dilakukan:
# Transiluminasi kepala bisa menunjukkan adanya cairan abnormal yang tertimbun di berbagai daerah di kepala
# CT scan kepala
# Pungsi lumbal dan pemeriksaan cairan serebrospinal
# Rontgen kepala (menunjukkan adanya penipisan dan pemisahan tulang tengkorak)
# Scan otak dengan radioisotop bisa menunjukkan adanya kelainan pada jalur cairan serebrospinal
# Arteriografi pembuluh darah otak.
# Ekoensefalogram (USG otak, menunjukkan adanya pelebaran ventrikel akibat hidrosefalus maupun perdarahan intraventrikuler).
Tujuan pengobatan adalah meminimalkan atau mencegah terjadinya kerusakan otak dengan cara memperbaiki aliran cairan serebrospinal.
Pembedahan adalah pengobatan utama pada hidrosefalus. Jika memungkinkan, sumber penyumbatan diangkat. Jika tidak memungkinkan, maka dibuat suatu shunt sehingga cairan serebrospinal tidak perlu melewati daerah yang tersumbat.
Shunting ke luar daerah otak bisa dilakukan ke atrium kanan jantung atau peritoneum.
Kauterisasi atau pengangkatan sebagian ventrikel yang menghasilkan cairan serebrospinal secara teoritis bisa mengurangi jumlah cairan serebrospinal yang dibentuk.
Jika ada tanda-tanda infeksi, segera diberikan antibiotik.
Jika terjadi infeksi yang berat, kemungkinan shunt harus dilepas.
Pemeriksaan lanjutan harus dilakukan secara rutin sepanjang hidup penderita untuk menilai tingkat perkembangan anak dan untuk mengobati setiap kelainan intelektual, saraf maupun fisik.
Jika tidak diobati, angka kematian mencapai 50-60%. Anak yang bertahan hidup akan mengalami kelainan intelektual, fisik dan saraf.
Prognosis untuk hidrosefalus yang diobati bervariasi, tergantung kepada penyebabnya. Jika anak bertahan hidup selama 1 tahun, maka hampir sepertiganya memiliki fungsi intelektual yang normal tetapi kelainan sarafnya tetap ada.
Hidrosefalus yang tidak disebabkan oleh infeksi memiliki prognosis yang terbaik, sedangkan jika penyebabnya adalah tumor maka prognosisnya paling buruk.
SINDROMA ARNOLD-CHIARI
Sindroma Arnold-Chiari adalah suatu kelainan pada pembentukan otak bagian bawah (batang otak).
Sindroma ini seringkali berhubungan dengan hidrosefalus.
Beberapa kelainan otak dapat diketahui sebelum bayi lahir, yaitu melalui pemeriksaan USG dan pemeriksaan cairan ketuban.
ANENSEFALUS
Anensefalus adalah suatu keadaan dimana sebagian besar tulang tengkorak dan otak tidak terbentuk.
Anensefalus adalah suatu kelainan tabung saraf (suatu kelainan yang terjadi pada awal perkembangan janin yang menyebabkan kerusakan pada jaringan pembentuk otak dan korda spinalis).
Anensefalus terjadi jika tabung saraf sebelah atas gagal menutup, tetapi penyebabnya yang pasti tidak diketahui.
Penelitian menunjukkan kemungkinan anensefalus berhubungan dengan racun di lingkungan juga kadar asam folat yang rendah dalam darah.
Anensefalus ditemukan pada 3,6-4,6 dari 10.000 bayi baru lahir.
Faktor resiko terjadinya anensefalus adalah:
- Riwayat anensefalus pada kehamilan sebelumnya
- Kadar asam folat yang rendah.
Resiko terjadinya anensefalus bisa dikurangi dengan cara meningkatkan asupan asam folat minimal 3 bulan sebelum hamil dan selama kehamilan bulan pertama.
Gejalanya berupa:
# Ibu : polihidramnion (cairan ketuban di dalam rahim terlalu banyak)
# Bayi
- tidak memiliki tulang tengkorak
- tidak memiliki otak (hemisfer serebri dan serebelum)
- kelainan pada gambaran wajah
- kelainan jantung.
Pemeriksaan yang biasa dilakukan adalah:
- Kadar asam lemak dalam serum ibu hamil
- Amniosentesis (untuk mengetahui adanya peningkatan kadar alfa-fetoprotein)
- Kadar alfa-fetoprotein meningkat (menunjukkan adanya kelainan tabung saraf)
- Kadar estriol pada air kemih ibu
- USG.
Bayi yang menderita anensefalus tidak akan bertahan, mereka lahir dalam keadaan meninggal atau akan meninggal dalam waktu beberapa hari setelah lahir.
MIKROSEFALUS
Mikrosefalus adalah suatu keadaan dimana ukuran kepala (lingkar puncak kepala) lebih kecil dari ukuran kepala rata-rata pada bayi berdasarkan umur dan jenis kelamin.
Dikatakan lebih kecil jika ukuran lingkar kepala kurang dari 42 cm atau lebih kecil dari standar deviasi 3 dibawah angka rata-rata.
Mikrosefalus seringkali terjadi akibat kegagalan pertumbuhan otak pada kecepatan yang normal. Berbagai keadaan dan penyakit yang mempengaruhi pertumbuhan otak bisa menyebabkan mikrosefalus.
Mikrosefalus seringkali berhubungan dengan keterbelakangan mental.
Mikrosefalus bisa terjadi setelah infeksi yang menyebabkan kerusakan pada otak pada bayi yang sangat muda (misalnya meningitis dan meningoensefalitis).
Penyebab utama:
# Sindroma Down
# Sindroma cri du chat
# Sindroma Seckel
# Sindroma Rubinstein-Taybi
# Trisomi 13
# Trisomi 18
# Sindroma Smith-Lemli-Opitz
# Sindroma Cornelia de Lange
Penyebab sekunder:
# Fenilketonuria pada ibu yang tidak terkontrol
# Keracunan metil merkuri
# Rubella kongenital
# Toksoplasmosis kongenital
# Sitomegalovirus kongenital
# Penyalahgunaan obat oleh ibu hamil
# Kekurangan gizi (malnutrisi).
Perawatan pada mikrosefalus tergantung kepada penyebabnya.
Bayi yang menderita mikrosefalus seringkali bisa bertahan hidup tetapi cenderung mengalami keterbelakangan mental, gangguan koordinasi otot dan kejang.
ENSEFALOKEL
Ensefalokel adalah suatu kelainan tabung saraf yang ditandai dengan adanya penonjolan meningens (selaput otak) dan otak yang berbentuk seperti kantung melalui suatu lubang pada tulang tengkorak.
Ensefalokel disebabkan oleh kegagalan penutupan tabung saraf selama perkembangan janin.
Gejalanya berupa:
- hidrosefalus
- kelumpuhan keempat anggota gerak (kuadriplegia spastik)
- gangguan perkembangan
- mikrosefalus
- gangguan penglihatan
- keterbelakangan mental dan pertumbuhan
- ataksia
- kejang.
Beberapa anak memiliki kecerdasan yang normal.
Ensefalokel seringkali disertai dengan kelainan kraniofasial atau kelainan otak lainnya.
Biasanya dilakukan pembedahan untuk mengembalikan jaringan otak yang menonjol ke dalam tulang tengkorak, membuang kantung dan memperbaiki kelainan kraniofasial yang terjadi.
Untuk hidrosefalus mungkin perlu dibuat suatu shunt. Pengobatan lainnya bersifat simtomatis dan suportif.
Prognosisnya tergantung kepada jaringan otak yang terkena, lokasi kantung dan kelainan otak yang menyertainya.
PORENSEFALUS
Porensefalus adalah suatu keadaan dimana pada hemisfer serebri ditemukan suatu kista atau rongga abnormal.
Porensefalus merupakan akibat dari kerusakan otak dan biasanya berhubungan dengan kelainan fungsi otak. Tetapi beberapa anak yang menderita porensefalus memiliki kecerdasan yang normal.
HIDRANENSEFALUS
Hidranensefalus adalah suatu keadaan dimana hemisfer serebri tidak ada dan digantikan oleh kantung-kantung yang berisi cairan serebrospinalis.
Ketika lahir, bayi tampak normal. Ukuran kepala dan refleks spontannya (misalnya refleks menghisap, menelan, menangis dan menggerakkan lengan dan tungkainya) tampak normal. Tetapi beberapa minggu kemudian, biasanya bayi menjadi rewel dan ketegangan ototnya meningkat.
Setelah berusia beberapa bulan, bisa terjadi kejang dan hidrosefalus. Gejala lainnya adalah:
- gangguan penglihatan
- gangguan pertumbuhan
- tuli
- buta
- kelumpuhan (kuadriplegi spastis)
- gangguan kecerdasan.
Hidranensefalus merupakan bentuk porensefalus yang ekstrim dan bisa disebabkan oleh infeksi pembuluh darah atau cedera yang terjadi pada saat usia kehamilan telah mencapai 12 minggu.
Diagnosisnya mungkin tertunda selama beberapa bulan karena perilaku awalnya relatif normal. Beberapa bayi menunjukkan kelainan pada saat lahir, yaitu berupa kejang, mioklonus (kejang atau kedutan otot atau sekelompok otot) dan gangguan pernafasan.
Tidak ada pengobatan yang pasti untuk hidranensefalus. Pengobatannya bersifat simtomatis dan suportif. Prognosis hidranensefalus adalah jelek, bayi biasanya meninggal sebelum berumur 1 tahun.
Hidrosefalus bisa diatasi dengan membuat shunt.
HIDROSEFALUS
Hidrosefalus adalah penimbunan cairan serebrospinal yang berlebihan di dalam otak.
Cairan serebrospinal dibuat di dalam otak dan biasanya beredar ke seluruh bagian otak, selaput otak serta kanalis spinalis, kemudian diserap ke dalam sistem peredaran darah.
Jika terjadi gangguan pada peredaran maupun penyerapan cairan serebrospinal, atau jika cairan yang dibentuk terlalu banyak, maka volume cairan di dalam otak menjadi lebih tinggi dari normal. Penimbunan cairan menyebabkan penekanan pada otak sehingga memaksa otak untuk mendorong tulang tengkorak atau merusak jaringan otak.
Gejalanya bervariasi, tergantung kepada penyebab dari penyumbatan aliran cairan serebrospinal dan luasnya kerusakan jaringan otak akibat hidrosefalus.
Pada bayi, cairan menumpuk pada sistem saraf pusat dan menyebabkan ubun-ubun menonjol serta kepala membesar. Kepala bisa membesar karena piringan tulang tengkorak belum sepenuhnya menutup. Tetapi, jika tulang tengkorak telah menutup (sekitar usia 5 tahun), maka tulang tengkorak tidak dapat membesar lagi.
Pada anak-anak, resiko terjadinya hidrosefalus ditemukan pada:
# Kelainan bawaan
# Tumor pada sistem saraf pusat
# Infeksi dalam kandungan
# Infeksi sistem saraf pusat pada bayi atau anak-anak (misalnya meningitis atau ensefalitis)
# Cedera pada proses kelahiran
# Cedera sebelum atau sesudah lahir (misalnya perdarahan subaraknoid).
Mielomeningokel adalah suatu penyakit dimana terjadi penutupan yan g tidak sempurna pada kolumna spinalis dan berhubungan erat dengan hidrosefalus.
Pada anak-anak yang lebih besar, resiko terjadinya hidrosefalus adalah:
# Riwayat kelainan bawaan
# Space-occupying lesions atau tumor otak maupun korda spinalis
# Infeksi sistem saraf pusat
# Perdarahan otak
# Trauma.
Gejala awal pada bayi:
- Kepala membesar
- Ubun-ubun menonjol dengan atau tanpa pembesaran kepala
- Sutura terpisah.
Gejala pada hidrosefalus lanjutan:
- Rewel, tidak dapat menahan emosi
- Kejang otot.
Gejala lanjut:
- Penurunan fungsi mental
- Gangguan perkembangan
- Penurunan pergerakan
- Gerakan menjadi lambat atau terhambat
- Tidak mau makan/menyusu
- Lemas, tidur terus
- Beser
- Menangis dengan nada tinggi, keras dan singkat
- Gangguan pertumbuhan.
Gejala pada bayi yang lebih tua dan anak-anak bervariasi, tergantung kepada luas kerusakan akibat penekanan terhadap otak. Gejalanya mirip dengan gejala hidrosefalus lanjutan pada anak-anak atau bisa berupa:
- Sakit kepala
- Muntah
- Gangguan penglihatan
- Juling
- Pergerakan mata yang tidak terkendali
- Hilangnya koordinasi
- Langkahnya tidak tegas
- Kelainan mental (misalnya psikosa).
Mengetuk tulang tengkorak secara perlahan dengan ujung jari tangan akan menunjukkan suara abnormal akibat penipisan dan pemisahan tulang-tulang tengkorak.
Vena pada tulang tengkorak tampak melebar. Lingkar kepala membesar atau kepala membesar hanya pada bagian tertentu (paling sering di daerah dahi). Mata tampak tertekan (gambaran sunset, dimana bagian putih mata terlihat diatas iris).
Pemeriksaan saraf menunjukkan adanya defisit neurologis fokal (gangguan fungsi saraf yang sifatnya lokal) dan refleks yang abnormal.
Pemeriksaan yang biasa dilakukan:
# Transiluminasi kepala bisa menunjukkan adanya cairan abnormal yang tertimbun di berbagai daerah di kepala
# CT scan kepala
# Pungsi lumbal dan pemeriksaan cairan serebrospinal
# Rontgen kepala (menunjukkan adanya penipisan dan pemisahan tulang tengkorak)
# Scan otak dengan radioisotop bisa menunjukkan adanya kelainan pada jalur cairan serebrospinal
# Arteriografi pembuluh darah otak.
# Ekoensefalogram (USG otak, menunjukkan adanya pelebaran ventrikel akibat hidrosefalus maupun perdarahan intraventrikuler).
Tujuan pengobatan adalah meminimalkan atau mencegah terjadinya kerusakan otak dengan cara memperbaiki aliran cairan serebrospinal.
Pembedahan adalah pengobatan utama pada hidrosefalus. Jika memungkinkan, sumber penyumbatan diangkat. Jika tidak memungkinkan, maka dibuat suatu shunt sehingga cairan serebrospinal tidak perlu melewati daerah yang tersumbat.
Shunting ke luar daerah otak bisa dilakukan ke atrium kanan jantung atau peritoneum.
Kauterisasi atau pengangkatan sebagian ventrikel yang menghasilkan cairan serebrospinal secara teoritis bisa mengurangi jumlah cairan serebrospinal yang dibentuk.
Jika ada tanda-tanda infeksi, segera diberikan antibiotik.
Jika terjadi infeksi yang berat, kemungkinan shunt harus dilepas.
Pemeriksaan lanjutan harus dilakukan secara rutin sepanjang hidup penderita untuk menilai tingkat perkembangan anak dan untuk mengobati setiap kelainan intelektual, saraf maupun fisik.
Jika tidak diobati, angka kematian mencapai 50-60%. Anak yang bertahan hidup akan mengalami kelainan intelektual, fisik dan saraf.
Prognosis untuk hidrosefalus yang diobati bervariasi, tergantung kepada penyebabnya. Jika anak bertahan hidup selama 1 tahun, maka hampir sepertiganya memiliki fungsi intelektual yang normal tetapi kelainan sarafnya tetap ada.
Hidrosefalus yang tidak disebabkan oleh infeksi memiliki prognosis yang terbaik, sedangkan jika penyebabnya adalah tumor maka prognosisnya paling buruk.
SINDROMA ARNOLD-CHIARI
Sindroma Arnold-Chiari adalah suatu kelainan pada pembentukan otak bagian bawah (batang otak).
Sindroma ini seringkali berhubungan dengan hidrosefalus.
Spina Bifida (Sumbing Tulang Belakang)
PENYEBAB
Resiko melahirkan anak dengan spina bifida berhubungan erat dengan kekurangan asam folat, terutama yang terjadi pada awal kehamilan.
Penonjolan dari korda spinalis dan meningens menyebabkan kerusakan pada korda spinalis dan akar saraf, sehingga terjadi penurunan atau gangguan fungsi pada bagian tubuh yang dipersarafi oleh saraf tersebut atau di bagian bawahnya.
Gejalanya tergantung kepada letak anatomis dari spina bifida. Kebanyakan terjadi di punggung bagian bawah, yaitu daerah lumbal atau sakral, karena penutupan vertebra di bagian ini terjadi paling akhir.
Kelainan bawaan lainnya yang juga ditemukan pada penderita spina bifida:
# Hidrosefalus
# Siringomielia
# Dislokasi pinggul.
GEJALA
Gejalanya bervariasi, tergantung kepada beratnya kerusakan pada korda spinalis dan akar saraf yang terkena.
Beberapa anak memiliki gejala ringan atau tanpa gejala; sedangkan yang lainnya mengalami kelumpuhan pada daerah yang dipersarafi oleh korda spinalis maupun akar saraf yang terkena.
Terdapat beberapa jenis spina bifida:
1. Spina bifida okulta : merupakan spina bifida yang paling ringan. Satu atau beberapa vertebra tidak terbentuk secara normal, tetapi korda spinalis dan selaputnya (meningens) tidak menonjol.
2. Meningokel : meningens menonjol melalui vertebra yang tidak utuh dan teraba sebagai suatu benjolan berisi cairan di bawah kulit.
3. Mielokel : jenis spina bifida yang paling berat, dimana korda spinalis menonjol dan kulit diatasnya tampak kasar dan merah.
Gejalanya berupa:
- penonjolan seperti kantung di punggung tengah sampai bawah pada bayi baru lahir
- jika disinari, kantung tersebut tidak tembus cahaya
- kelumpuhan/kelemahan pada pinggul, tungkai atau kaki
- penurunan sensasi
- inkontinensia uri (beser) maupun inkontinensia tinja
- korda spinalis yang terkena rentan terhadap infeksi (meningitis).
Gejala pada spina bifida okulta:
- seberkas rambut pada daerah sakral (panggul bagian belakang)
- lekukan pada daerah sakrum.
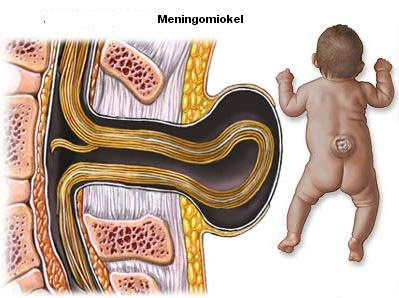
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan fisik.
Pada trimester pertama, wanita hamil menjalani pemeriksaan darah yang disebut triple screen. Tes ini merupakan tes penyaringan untuk spina bifida, sindroma Down dan kelainan bawaan lainnya.
85% wanita yang mengandung bayi dengan spina bifida, akan memiliki kadar serum alfa fetoprotein yang tinggi. Tes ini memiliki angka positif palsu yang tinggi, karena itu jika hasilnya positif, perlu dilakukan pemeriksaan lanjutan untuk memperkuat diagnosis. Dilakukan USG yang biasanya dapat menemukan adanya spina bifida.
Kadang dilakukan amniosentesis (analisa cairan ketuban).
Setelah bayi lahir, dilakukan pemeriksaan berikut:
- Rontgen tulang belakang untuk menentukan luas dan lokasi kelainan.
- USG tulang belakang bisa menunjukkan adanya kelainan pda korda spinalis maupun vertebra
- CT scan atau MRI tulang belakang kadang dilakukan untuk menentukan lokasi dan luasnya kelainan.
PENGOBATAN
Tujuan dari pengobatan awal adalah:
- mengurangi kerusakan saraf akibat spina bifida
- meminimalkan komplikasi (misalnya infeksi)
- membantu keluarga dalam menghadapi kelainan ini.
Pembedahan dilakukan untuk menutup lubang yang terbentuk dan untuk mengobati hidrosefalus, kelainan ginjal dan kandung kemih serta kelainan bentuk fisik yang sering menyertai spina bifida.
Terapi fisik dilakukan agar pergerakan sendi tetap terjaga dan untuk memperkuat fungsi otot.
Untuk mengobati atau mencegah meningitis, infeksi saluran kemih dan infeksi lainnya, diberikan antibiotik.
Untuk membantu memperlancar aliran air kemih bisa dilakukan penekanan lembut diatas kandung kemih. Pada kasus yang berat kadang harus dilakukan pemasangan kateter.
Diet kaya serat dan program pelatihan buang air besar bisa membantu memperbaiki fungsi saluran pencernaan.
Untuk mengatasi gejala muskuloskeletal (otot dan kerangka tubuh) perlu campur tangan dari ortopedi (bedah tulang) maupun terapi fisik. Kelainan saraf lainnya diobati sesuai dengan jenis dan luasnya gangguan fungsi yang terjadi.
Kadang pembedahan shunting untuk memperbaiki hidrosefalus akan menyebabkan berkurangnya mielomeningokel secara spontan .
PENCEGAHAN
Resiko terjadinya spina bifida bisa dikurangi dengan mengkonsumsi asam folat.
Kekurangan asam folat pada seorang wanita harus dikoreksi sebelum wanita tersebut hamil, karena kelainan ini terjadi sangat dini.
Kepada wanita yang berencana untuk hamil dianjurkan untuk mengkonsumsi asam folat sebanyak 0,4 mg/hari. Kebutuhan asam folat pada wanita hamil adalah 1 mg/hari.
Kelainan Ginjal & Saluran Kemih Bawaan
Kelainan bawaan pada ginjal dan saluran kemiih lebih sering ditemukan daripada kelainan bawaan pada bagian tubuh lainnya.
Kelainan bawaan yang menyumbat aliran air kemih menyebabkan air kemih tertahan dan hal ini bisa menyebabkan infeksi atau pembentukan batu ginjal.
Suatu kelainan bawaan pada sistem kemih-kelamin bisa menyebabkan gangguan fungsi ginjal atau menyebabkan kelainan fungsi seksual maupun kemandulan di kemudian hari.
KELAINAN GINJAL & URETER
Pada saat pembentukan ginjal bisa terjadi sejumlah kelainan:
Kelainan bawaan yang menyumbat aliran air kemih menyebabkan air kemih tertahan dan hal ini bisa menyebabkan infeksi atau pembentukan batu ginjal.
Suatu kelainan bawaan pada sistem kemih-kelamin bisa menyebabkan gangguan fungsi ginjal atau menyebabkan kelainan fungsi seksual maupun kemandulan di kemudian hari.
KELAINAN GINJAL & URETER
Pada saat pembentukan ginjal bisa terjadi sejumlah kelainan:
# Ektopia : ginjal tidak terletak pada tempat yang seharusnya
# Malrotasi : ginjal berada pada posisi yang salah
# Horseshoe kidney : kedua ginjal bersatu
# Agenesis ginjal : ginjal tidak terbentuk
# Sindroma Potter : kedua ginjal tidak terbentuk
# Penyakit ginjal polikista : ginjal mengandung banyak kista.
Kelainan yang mungkin ditemukan pada ureter (saluran kemih yang menghubungkan ginjal dengan kandung kemih):
# Ekstra ureter
# Misplaced ureter
# Ureter yang melebar atau menyempit.
Air kemih bisa mengalir balik dari kandung kemih ke dalam ureter yang abnormal, sehingga mudah terjadi infeksi ginjal (pielonefritis).
Ureter yang menyempit bisa menghalangi aliran air kemih dari ginjal ke kandung kemih dan bisa menyebabkan ginjal membesar (hidronefrosis) serta menyebabkan kerusakan ginjal.
Horseshoe Kidney
Pada horseshoe kidney, ginjal menyatu pada bagian bawahnya sehingga bentuknya menyerupai tapal kuda.
Kelainan ini tidak menimbulkan gejala atau masalah dan seringkali tidak terdeteksi. Ginjal tapal kuda mungkin ditemukan secara tidak sengaja pada pemeriksaan rontgen atau USG di daerah ginjal yang dilakukan untuk keperluan lain.
Pada beberapa kasus, ginjal tapal kuda bisa menyebabkan gangguan pada pengaliran air kemih ke dalam ureter. Hal ini akan menyebabkan meningkatnya resiko infeksi ginjal dan kerusakan fungsi ginjal.
Jika timbul gangguan, maka keadaan ini bisa diperbaiki melalui pembedahan.

Ginjal Agenesis
Sekitar 1 diantara 1.500 bayi terlahir hanya dengan satu ginjal. Ginjal ini biasanya lebih besar dari normal.
Dengan satu ginjal, seseorang masih bisa menjalani kehidupan yang normal. Tetapi ada 3 hal penting yang dapat membantu menjamin kesehatan ginjal:
- Katakan pada setiap dokter yang anda kunjungi bahwa anda hanya memiliki satu ginjal.
- Untuk mengurangi kemungkinan terbentuknya batu ginjal, minum air putih sebanyak mungkin. Batu dapat merusak ginjal.
- Lakukan pemeriksaan kesehatan secara teratur.
Sindroma Potter
Sindroma Potter dan Fenotip Potter adalah suatu keadaan kompleks yang berhubungan dengan gagal ginjal bawaan dan berhubungan dengan oligohidramnion (cairan ketuban yang sedikit).
Fenotip Potter digambarkan sebagai suatu keadaan khas pada bayi baru lahir, dimana cairan ketubannya sangat sedikit atau tidak ada. Oligohidramnion menyebabkan bayi tidak memiliki bantalan terhadap dinding rahim. Tekanan dari dinding rahim menyebabkan gambaran wajah yang khas (wajah Potter). Selain itu, karena ruang di dalam rahim sempit, maka anggota gerak tubuh menjadi abnormal atau mengalami kontraktur dan terpaku pada posisi abnormal.
Oligohidramnion juga menyebabkan terhentinya perkembangan paru-paru (paru-paru hipoplastik), sehingga pada saat lahir, paru-paru tidak berfungsi sebagaimana mestinya.
Pada sindroma Potter, kelainan yang utama adalah gagal ginjal bawaan, baik karena kegagalan pembentukan ginjal (agenesis ginjal bilateral) maupun karena penyakit lain pada ginjal yang menyebabkan ginjal gagal berfungsi.
Dalam keadaan normal, ginjal membentuk cairan ketuban (sebagai air kemih) dan tidak adanya cairan ketuban menyebabkan gambaran yang khas dari sindroma Potter.
Gejalanya berupa:
- Wajah Potter (kedua mata terpisah jauh, terdapat lipatan epikantus, pangkal hidung yang lebar, telinga yang rendah dan dagu yang tertarik ke belakang)
- Tidak terbentuk air kemih
- Gawat pernafasan,
Diagnosis ditegakkan berdasarkan adanya fenotip Potter, oligohidramnion, paru-paru yang kaku dan kelainan sistem saluran kemih-kelamin.
Pemeriksaan yang biasa dilakukan:
- USG ibu (menunjukkan oligohidramnion serta tidak adanya ginjal janin atau ginjal yang sangat abnormal)
- Rontgen perut bayi
- Rontgen paru-paru bayi
- Analisa gas darah.
Pengobatan ditujukan untuk memperbaiki penyumbatan pada saluran kemih.
KELAINAN KANDUNG KEMIH
Sejumlah kelainan bawaan yang bisa ditemukan pada kandung kemih:
# Ekstrofi : kandung kemih tidak terbentuk secara sempurna sehingga terbuka ke permukaan perut.
# Kelainan pada dinding kandung kemih, yaitu adanya kantung divertikula) yang memungkinkan aliran kemih terhenti dan meningkatkan resiko terjadinya infeksi saluran kemih.
# Penyempitan pada lubang kandung kemih yang tersambung ke uretra bisa menyebabkan pengosongan kandung kemih yang tidak sempurna. Aliran kemih menjadi lambat dan bisa terjadi infeksi.
Kelainan kandung kemih bisa diatasi dengan pembedahan.
KELAINAN URETRA
Uretra adalah saluran yang mengalirkan air kemih keluar tubuh. Uretra bisa abnormal atau tidak terbentuk.
Pada anak laki-laki bisa ditemukan kelainan berikut:
# Hipospadia : lubang uretra berada pada penis bagian bawah
# Epispadia : uretra pada penis terbuka dan tidak tertutup seperti halnya sebuah tabung.
Baik pada anak perempuan maupun anak laki-laki, penyempitan uretra bisa menyumbat aliran air kemih.
Kelainan pada uretra bisa diperbaiki melalui pembedahan.
Hipospadia
Hipospadia adalah suatu keadaan dimana lubang uretra terdapat di penis bagian bawah, bukan di ujung penis.
Hipospadia merupakan kelainan bawaan yang terjadi pada 3 diantara 1.000 bayi baru lahir.
Beratnya hipospadia bervariasi, kebanyakan lubang uretra terletak di dekat ujung penis, yaitu pada glans penis.
Bentuk hipospadia yang lebih berat terjadi jika lubang uretra terdapat di tengah batang penis atau pada pangkal penis, dan kadang pada skrotum (kantung zakar) atau di bawah skrotum. Kelainan ini seringkali berhubungan dengan kordi, yaitu suatu jaringan fibrosa yang kencang, yang menyebabkan penis melengkung ke bawah pada saat ereksi.
Gejalanya adalah:
- Lubang penis tidak terdapat di ujung penis, tetapi berada di bawah atau di dasar penis
- Penis melengkung ke bawah
- Penis tampak seperti berkerudung karena adanya kelainan pada kulit depan penis
- Jika berkemih, anak harus duduk.
Diagnosis ditegakkan berdasarkan pemeriksaan fisik.
Jika hipospadia terdapat di pangkal penis, mungkin perlu dilakukan pemeriksaan radiologis untuk memeriksa kelainan bawaan lainnya.
Bayi yang menderita hipospadia sebaiknya tidak disunat. Kulit depan penis dibiarkan untuk digunakan pada pembedahan nanti.
Rangkaian pembedahan biasanya telah selesai dilakukan sebelum anak mulai sekolah. Pada saat ini, perbaikan hipospadia dianjurkan dilakukan sebelum anak berumur 18 bulan.
Jika tidak diobati, mungkin akan terjadi kesulitan dalam pelatihan buang air pada anak dan pada saat dewasa nanti, mungkin akan terjadi gangguan dalam melakukan hubungan seksual.

Epispadia
Epispadia adalah suatu kelainan bawaan pada bayi laki-laki, dimana lubang uretra terdapat di bagian punggung penis atau uretra tidak berbentuk tabung, tetapi terbuka.
Terdapat 3 jenis epispadia:
# Lubang uretra terdapat di puncak kepala penis
# Seluruh uretra terbuka di sepanjang penis
# Seluruh uretra terbuka dan lubang kandung kemih terdapat pada dinding perut.
Gejalanya adalah:
- Lubang uretra terdapat di punggung penis
- Lubang uretra terdapat di sepanjang punggung penis.
Untuk menilai beratnya epispadia, dilakukan pemeriksaan berikut:
- Radiologis (IVP)
- USG sistem kemih-kelamin.
Epispadia biasanya diperbaiki melalui pembedahan.
# Malrotasi : ginjal berada pada posisi yang salah
# Horseshoe kidney : kedua ginjal bersatu
# Agenesis ginjal : ginjal tidak terbentuk
# Sindroma Potter : kedua ginjal tidak terbentuk
# Penyakit ginjal polikista : ginjal mengandung banyak kista.
Kelainan yang mungkin ditemukan pada ureter (saluran kemih yang menghubungkan ginjal dengan kandung kemih):
# Ekstra ureter
# Misplaced ureter
# Ureter yang melebar atau menyempit.
Air kemih bisa mengalir balik dari kandung kemih ke dalam ureter yang abnormal, sehingga mudah terjadi infeksi ginjal (pielonefritis).
Ureter yang menyempit bisa menghalangi aliran air kemih dari ginjal ke kandung kemih dan bisa menyebabkan ginjal membesar (hidronefrosis) serta menyebabkan kerusakan ginjal.
Horseshoe Kidney
Pada horseshoe kidney, ginjal menyatu pada bagian bawahnya sehingga bentuknya menyerupai tapal kuda.
Kelainan ini tidak menimbulkan gejala atau masalah dan seringkali tidak terdeteksi. Ginjal tapal kuda mungkin ditemukan secara tidak sengaja pada pemeriksaan rontgen atau USG di daerah ginjal yang dilakukan untuk keperluan lain.
Pada beberapa kasus, ginjal tapal kuda bisa menyebabkan gangguan pada pengaliran air kemih ke dalam ureter. Hal ini akan menyebabkan meningkatnya resiko infeksi ginjal dan kerusakan fungsi ginjal.
Jika timbul gangguan, maka keadaan ini bisa diperbaiki melalui pembedahan.
Ginjal Agenesis
Sekitar 1 diantara 1.500 bayi terlahir hanya dengan satu ginjal. Ginjal ini biasanya lebih besar dari normal.
Dengan satu ginjal, seseorang masih bisa menjalani kehidupan yang normal. Tetapi ada 3 hal penting yang dapat membantu menjamin kesehatan ginjal:
- Katakan pada setiap dokter yang anda kunjungi bahwa anda hanya memiliki satu ginjal.
- Untuk mengurangi kemungkinan terbentuknya batu ginjal, minum air putih sebanyak mungkin. Batu dapat merusak ginjal.
- Lakukan pemeriksaan kesehatan secara teratur.
Sindroma Potter
Sindroma Potter dan Fenotip Potter adalah suatu keadaan kompleks yang berhubungan dengan gagal ginjal bawaan dan berhubungan dengan oligohidramnion (cairan ketuban yang sedikit).
Fenotip Potter digambarkan sebagai suatu keadaan khas pada bayi baru lahir, dimana cairan ketubannya sangat sedikit atau tidak ada. Oligohidramnion menyebabkan bayi tidak memiliki bantalan terhadap dinding rahim. Tekanan dari dinding rahim menyebabkan gambaran wajah yang khas (wajah Potter). Selain itu, karena ruang di dalam rahim sempit, maka anggota gerak tubuh menjadi abnormal atau mengalami kontraktur dan terpaku pada posisi abnormal.
Oligohidramnion juga menyebabkan terhentinya perkembangan paru-paru (paru-paru hipoplastik), sehingga pada saat lahir, paru-paru tidak berfungsi sebagaimana mestinya.
Pada sindroma Potter, kelainan yang utama adalah gagal ginjal bawaan, baik karena kegagalan pembentukan ginjal (agenesis ginjal bilateral) maupun karena penyakit lain pada ginjal yang menyebabkan ginjal gagal berfungsi.
Dalam keadaan normal, ginjal membentuk cairan ketuban (sebagai air kemih) dan tidak adanya cairan ketuban menyebabkan gambaran yang khas dari sindroma Potter.
Gejalanya berupa:
- Wajah Potter (kedua mata terpisah jauh, terdapat lipatan epikantus, pangkal hidung yang lebar, telinga yang rendah dan dagu yang tertarik ke belakang)
- Tidak terbentuk air kemih
- Gawat pernafasan,
Diagnosis ditegakkan berdasarkan adanya fenotip Potter, oligohidramnion, paru-paru yang kaku dan kelainan sistem saluran kemih-kelamin.
Pemeriksaan yang biasa dilakukan:
- USG ibu (menunjukkan oligohidramnion serta tidak adanya ginjal janin atau ginjal yang sangat abnormal)
- Rontgen perut bayi
- Rontgen paru-paru bayi
- Analisa gas darah.
Pengobatan ditujukan untuk memperbaiki penyumbatan pada saluran kemih.
KELAINAN KANDUNG KEMIH
Sejumlah kelainan bawaan yang bisa ditemukan pada kandung kemih:
# Ekstrofi : kandung kemih tidak terbentuk secara sempurna sehingga terbuka ke permukaan perut.
# Kelainan pada dinding kandung kemih, yaitu adanya kantung divertikula) yang memungkinkan aliran kemih terhenti dan meningkatkan resiko terjadinya infeksi saluran kemih.
# Penyempitan pada lubang kandung kemih yang tersambung ke uretra bisa menyebabkan pengosongan kandung kemih yang tidak sempurna. Aliran kemih menjadi lambat dan bisa terjadi infeksi.
Kelainan kandung kemih bisa diatasi dengan pembedahan.
KELAINAN URETRA
Uretra adalah saluran yang mengalirkan air kemih keluar tubuh. Uretra bisa abnormal atau tidak terbentuk.
Pada anak laki-laki bisa ditemukan kelainan berikut:
# Hipospadia : lubang uretra berada pada penis bagian bawah
# Epispadia : uretra pada penis terbuka dan tidak tertutup seperti halnya sebuah tabung.
Baik pada anak perempuan maupun anak laki-laki, penyempitan uretra bisa menyumbat aliran air kemih.
Kelainan pada uretra bisa diperbaiki melalui pembedahan.
Hipospadia
Hipospadia adalah suatu keadaan dimana lubang uretra terdapat di penis bagian bawah, bukan di ujung penis.
Hipospadia merupakan kelainan bawaan yang terjadi pada 3 diantara 1.000 bayi baru lahir.
Beratnya hipospadia bervariasi, kebanyakan lubang uretra terletak di dekat ujung penis, yaitu pada glans penis.
Bentuk hipospadia yang lebih berat terjadi jika lubang uretra terdapat di tengah batang penis atau pada pangkal penis, dan kadang pada skrotum (kantung zakar) atau di bawah skrotum. Kelainan ini seringkali berhubungan dengan kordi, yaitu suatu jaringan fibrosa yang kencang, yang menyebabkan penis melengkung ke bawah pada saat ereksi.
Gejalanya adalah:
- Lubang penis tidak terdapat di ujung penis, tetapi berada di bawah atau di dasar penis
- Penis melengkung ke bawah
- Penis tampak seperti berkerudung karena adanya kelainan pada kulit depan penis
- Jika berkemih, anak harus duduk.
Diagnosis ditegakkan berdasarkan pemeriksaan fisik.
Jika hipospadia terdapat di pangkal penis, mungkin perlu dilakukan pemeriksaan radiologis untuk memeriksa kelainan bawaan lainnya.
Bayi yang menderita hipospadia sebaiknya tidak disunat. Kulit depan penis dibiarkan untuk digunakan pada pembedahan nanti.
Rangkaian pembedahan biasanya telah selesai dilakukan sebelum anak mulai sekolah. Pada saat ini, perbaikan hipospadia dianjurkan dilakukan sebelum anak berumur 18 bulan.
Jika tidak diobati, mungkin akan terjadi kesulitan dalam pelatihan buang air pada anak dan pada saat dewasa nanti, mungkin akan terjadi gangguan dalam melakukan hubungan seksual.
Epispadia
Epispadia adalah suatu kelainan bawaan pada bayi laki-laki, dimana lubang uretra terdapat di bagian punggung penis atau uretra tidak berbentuk tabung, tetapi terbuka.
Terdapat 3 jenis epispadia:
# Lubang uretra terdapat di puncak kepala penis
# Seluruh uretra terbuka di sepanjang penis
# Seluruh uretra terbuka dan lubang kandung kemih terdapat pada dinding perut.
Gejalanya adalah:
- Lubang uretra terdapat di punggung penis
- Lubang uretra terdapat di sepanjang punggung penis.
Untuk menilai beratnya epispadia, dilakukan pemeriksaan berikut:
- Radiologis (IVP)
- USG sistem kemih-kelamin.
Epispadia biasanya diperbaiki melalui pembedahan.
Intersex States
PENYEBAB
Berbagai keadaan yang bisa menyebabkan terjadinya genitalis ambigus dan hermafroditisme:
# Hiperplasia adrenal kongenital
# Pemaparan progestin atau androgen pada janin
# Sindroma feminisasi testikuler
# Disgenesis gonad XY
# Agenesis gonad XY
# Kelainan kromosom
# Kriptoftalmos
# Smith-Lemli-Opitz
# Sindroma 4p
# Sindroma 13q.

GEJALA
Pada saat lahir, bayi memiliki alat kelamin yang tidak jelas.
Hermafrodit sejati memiliki jaringan ovarium (indung telur) dan testis (buah zakar) serta organ reproduksi bagian dalam pria dan wanita. Tetapi hal ini jarang terjadi.
Anak dengan genitalis ambigus adalah pseudohermafrodit, dimana alat kelamin luarnya tidak jelas, dengan alat reproduksi bagian dalam berupa ovarium atau testis.
Seorang pseudohermafrodit wanita, secara genetis adalah wanita yang normal (memiliki 2 kromosom X), yang lahir dengan alat kelamin yang menyerupai penis yang kecil. Alat reproduksi bagian dalamnya adalah wanita.
Pseudohermafroditisme pada wanita terjadi akibat pemaparan terhadap kadar hormon pria yang tinggi selama berada dalam kandungan.
Biasanya janin memiliki kelenjar adrenal yang besar (sindroma adrenogenital) yang terlalu banyak menghasilkan hormon pria atau terdapat kekurangan enzim sehingga hormon pria tidak dapat diubah menjadi hormon wanita.
Kadang hormon pria dari darah ibu masuk ke dalam plasenta (ari-ari); misalnya jika ibu telah mengkonsumsi progesteron untuk mencegah keguguran atau ibu menderita tumor yang menghasilkan hormon.
Seorang pseudohermafrodit pria secara genetis adalah pria (dengan 1 kromosom X dan 1 kromosom Y), yang terlahir tanpa penis atau penisnya sangat kecil.
Tubuhnya gagal membentuk hormon pria atau melawan hormon yang dihasilkan (sindroma resistensi androgen).
DIAGNOSA
Diagnosis ditegakkan berdasarkan hasil pemeriksaan fisik.
Pemeriksaan lainnya yang biasa dilakukan:
# Analisa kromosom
# Kadar hormon (misalnya kadar testosteron)
# Pemeriksaan endoskopi (untuk mengetahui ada atau tidaknya vagina atau serviks).
PENGOBATAN
Untuk merekonstruksi alat kelamin luar, dilakukan pembedahan korektif.
Secara umum, lebih mudah untuk merekonstruksi kelamin wanita daripada kelamin pria.
Pembedahan untuk memperbaiki keadaan ini biasanya dilakukan pada saat mendekati masa pubertas.
Sindroma 4p
PENYEBAB
Penyebabnya adalah hilangnya bahan pada ujung lengan pendek kromosom 4.
GEJALA
Pada saat lahir, bayi telah menunjukkan gambaran wajah dismorfik:
- Mikrosefalus (kepala kecil)
- Kelainan pada garis tengah tulang tengkorak
- Hemangioma (tumor jinak pembuluh darah) di dahi
- Hipertelorisme (jarak antara kedua mata sangat pendek)
- Garis palpebra sipit ke bawah
- Epikantus (lipatan kulit pada kelopak mata)
- Strabismus (mata juling)
- Kolobomata (celah pada mata dan jaringan di sekitarnya)
- Letak telinga lebih rendah dan lipatan telinganya lebih sedikit
- Lekukan kulit di depan telinga
- Hidungnya lebar atau seperti paruh
- Celah bibir/celah langit-langit
- Mulutnya seperti ikan gurame
- Mikrognatia (lidah kecil).
Kelainan saraf:
- Keterbelakangan mental
- Keterbelakangan psikomotorik yang berat
- Pada masa bayi, tangisannya lemah
- Kejang.
Kelainan ginjal:
- Hipospadia
- Derivat Mullerian hipoplastik.
Gejala lainnya:
- Berat badan lahir rendah (rata-rata dibawah 2.105 gram)
- Pertumbuhannya lambat.
DIAGNOSA
Diagnosis ditegakkan berdasarkan gejala dan hasil pemeriksaan rontgen kerangka tubuh, yang menunjukkan danya penundaan proses osifikasi tulang karpal (pergelangan tangan) dan panggul.
Diagnosis prenatal (sebelum bayi lahir) ditunjukkan dengan adanya gangguan pertumbuhan di dalam rahim (IUGR, intrauterine growth retardation) disertai kelainan pada garis tengah dan mulut yang menghadap ke bawah.
PENGOBATAN
Tidak ada pengobatan khusus untuk sindroma 4p.
Sindroma Cri Du Chat : Tangisan Anak Seperti Suara Seekor Kucing
Penamaan sindroma ini didasarkan kepada tangisan bayi yang bernada tinggi dan terdengar seperti suara seekor kucing. Tangisan ini terdengar segera setelah bayi lahir dan berlangsung selama beberapa minggu, kemudian menghilang.
Sindroma ini ditemukan pada 1 diantara 20.000 dan 1 diantara 50.000 bayi.

PENYEBAB
Sindroma ini terjadi karena adanya penghapusan informasi pada kromosom 5.
Penyebab terjadinya penghapusan ini tidak diketahui, tetapi pada sebagian besar kasus, diperkirakan penyebabnya adalah hilangnya 1 keping kromosom 5 pada saat pembentukan sel telur atau sperma. Kasus lainnya terjadi karena salah satu orang tua membawa kromosom 5 yang telah mengalami translokasi (penyusunan ulang).
GEJALA
Gejalanya berupa:
- Tangisan bernada tinggi seperti suara kucing
- Berat badan lahir yang rendah dan pertumbuhan yang lambat
- Bayi tampak lemas
- Kepalanya kecil (mikrosefalus)
- Wajah asimetris dan mulutnya tidak dapat menutup rapat
- Hidungnya lebar
- Lehernya pendek
- Beberapa bayi memiliki wajah yang bundar (moon face)
- Hipertelorisme (kedua mata terpisah jauh)
- Fissura palpebra (mata sipit ke bawah)
- Mikrognatia (rahang kecil)
- Letak telinga lebih rendah (mungkin bentuknya juga abnormal)
- Skin tag di depan telinga
- Di sela jari kaki maupun tangan terdapat kulit tambahan (seperti selaput) atau jari-jarinya menyatu - Simian crease (garis tangan pada telapak tangan hanya satu)
- Keterbelakangan mental
- Perkembangan kemampuan motoriknya lambat atau tidak lengkap
- Sering disertai kelainan jantung.
DIAGNOSA
Selain gejala-gejala tersebut, pada pemeriksaan fisik juga bisa ditemukan:
- Hernia inguinalis
- Diastasis rekti (otot-otot perut terpisah)
- Otot kendur
- Lipatan epikantus (lipatan pada kulit di sudut mata sebelah dalam)
- Lipatan telinga yang tidak lengkap atau abnormal.
Analisa kromosom menunjukkan hilangnya lengan pendek pada kromosom 5. Analisa FISH menunjukkan adanya bagian kromosom 5 yang hilang.
Rontgen tulang tengkorak lateral menunjukkan sudut yang abnormal pada dasar tulang tengkorak.
PENGOBATAN
Tidak ada pengobatan khusus untuk sindroma cri du chat.
Kekurangan Gizi (Malnutrisi)
Kekurangan gizi bisa disebabkan oleh kurangnya asupan gizi atau ketidakmampuan tubuh untuk menyerap atau memetabolisir zat gizi.
Kekurangan gizi bisa terjadi ketika kebutuhan akan zat-zat gizi yang penting meningkat, misalnya pada saat mengalami stres, infeksi, cedera atau penyakit.
Kekurangan Kalori Protein (KKP) merupakan salah satu bentuk kekurangan gizi yang paling serius. KKP terjadi pada bayi akibat tidak adekuatnya masa menyusui ataupun masa menyapih.
KKP relatif sering ditemukan di negara-negara berkembang; di negara maju, bentuk KKP yang lebih ringan ditemukan pada keluarga misikin.
Sebagai bagian dari perawatan anak rutin, dokter akan menanyakan kepada orang tua maupun anak mengenai makanan dan intoleransi terhadap makanan serta memeriksa anak untuk mencari tanda-tanda dari kekurangan gizi atau kelainan yang mempengaruhi keadaan gizi (misalnya malabsorbsi, penyakit ginjal, diare, kelainan metabolik, kelainan genetik).
Pertumbuhan anak dinilai melalui pengukuran tinggi badan dan berat badan dan membandingkannya dengan grafik pertumbuhan yang normal.
Jika diduga terjadi malnutrisi, untuk memperkuat diagnosis bisa dilakukan pemeriksaan darah atau air kemih guna mengukur kadar zat gizi.
KEKURANGAN VITAMIN E
Kekurangan vitamin E relatif sering dijumpai pada bayi prematur karena penghantaran vitamin-vitamin yang larut dalam lemak oleh plasenta tidak berlangsung terlalu baik dan keadaan ini semakin diperburuk oleh prematuritas bayi.
Susu formula yang kaya akan asam lemak tak jenuh ganda menyebabkan meningkatnya kebutuhan akan vitamin E, terutama pada bayi prematur yang tidak mampu menyerap vitamin E.
Kekurangan vitamin E juga bisa terjadi pada anak-anak yang menderita penyakit yang menyebabkan gangguan penyerapan lemak, seperti fibrosis kistik dan kelainan genetik tertentu.
Pemberian zat besi yang berlebihan juga dapat menyebabkan kekurangan vitamin E.
Bayi prematur yang menderita kekurangan vitamin E, pada usia 6-10 minggu bisa mengalami kelemahan otot disertai anemia hemolitik, akibat berkurangnya kadar vitamin E dalam darah.
Hal ini bisa diatasi dengan pemberian vitamin E.
Kekurangan vitamin E memegang peran penting dalam terjadinya retinopati pada prematuritas, yaitu suatu kelainan mata yang akan semakin memburuk jika bayi terkena oksigen kadar tinggi di dalam inkubator.
Anak-nak yang menderita malabsorbsi usus bisa mengalami kekurangan vitamin E yang berat, yang menyebabkan sejumlah gejala neurologis (saraf), seperti berkurangnya refleks, kesulitan dalam berjalan, penglihatan ganda, hilangnya sensasi posisi dan kelemahan otot.
Gejala-gejala tersebut akan memburuk secara progresif, tetapi dapat diatasi dengan pengobatan.
KEKURANGAN VITAMIN K
Pada bayi baru lahir, bentuk kekurangan vitamin K yang sering ditemukan adalah penyakit hemoragik pada bayi baru lahir. Penyakit ini terjadi karena:
# Plasenta tidak terlalu baik dalam menghantarkan lemak (termasuk vitamin yang larut dalam lemak)
# Hati bayi yang baru lahir masih kurang matang untuk menghasilkan sejumlah protrombin (salah satu faktor pembekuan darah)
# Air susu ibu mengandung sedikit vitamin K, yaitu hanya 1-3 mikrogram/L, sedangkan susu sapi mengandung 5-10 mikrogram/L
# Pada beberapa hari pertama kehidupan bayi, di dalam ususnya belum ditemukan bakteri penghasil vitamin K.
Penyakit hemoragik pada bayi baru lahir biasanya terjadi pada hari ke 1-7.
Gejalanya berupa perdarahan di dalam kulit, di dalam lambung atau di dalam dada. Pada kasus yang sangat berat, perdarahan bisa terjadi di dalam otak.
Penyakit hemoragik lanjut timbul pada usia 1-3 bulan dan menyebabkan gejala yang sama dengan penyakit hemoragik pada bayi baru lahir.
Penyakit ini biasanya berhubungan dengan malabsorbsi atau penyakit hati.
Angka kejadian kedua penyakit hemoragik tersebut meningkat pada bayi-bayi yang ketika masih berada dalam kandungan, ibunya mengkonsumsi:
- obat anti-kejang hidantoin (misalnya phenitoin)
- antibiotik cephalosporin
- antikoagulan kumarin (misalnya warfarin).
Untuk mencegah terjadinya penyakit hemoragik pada bayi baru lahir, dianjurkan untuk memberikan suntikan vitamin K melalui otot dalam waktu 1 jam setelah bayi lahir. Pemberian melalui mulut tidak dianjurkan karena penyerapannya bervariasi dan keberadaanya di dalam tubuh tidak dapat diramalkan.
SKURVI INFANTIL
Skurvi Infantil adalah suatu keadaan yang disebabkan oleh tidak adekuatnya asupan vitamin C (asam askorbat), karena pemakaian susu formula yang hanya mengandung sedikit vitamin C.
Penyakit ini biasanya timbul pada usia 6-12 bulan.
Gejala awal berupa rewel, nafsu makan yang buruk dan berat badan tidak bertambah.
Jika digerakkan, bayi akan menjerit dan tidak mau menggerakkan tungkainya karena nyeri akibat perdarahan di bawah lapisan tipis pada jaringan pembungkus tulang.
Pada anak-anak yang lebih besar, perdarahan bisa terjadi di bawah kulit; gusi di sekeliling gigi yang sedang tumbuh juga mudah berdarah.
Vitamin C penting untuk pembentukan jaringan ikat (jaringan yang mengikat struktur tubuh), skurvi bisa menyebabkan kelainan pada tulang rusuk dan pada tulang panjang tungkai.
Pada tulang rusuk, persambungan diantara tulang dan tulang rawan melebar, membentuk sederetan benjolan yang disebut tasbih skorbutik.
Skurvi juga menyebabkan terganggunya proses penyembuhan luka.
Skurvi bisa dicegah dengan memberikan vitamin C yang adekuat, sumber yang sangat baik adalah buah-buahan dan jus asam (lemon, jeruk).
Kepada bayi yang diberi susu formula, sebaiknya diberikan vitamin C sebanyak 35 mgr/hari (sama dengan 85 gram jus jeruk/lemon).
Ibu yang menyusui sebaiknya mengkonsumsi vitamin C sebanyak 100 mg/hari.
Untuk mengobati skurvi, diberikan vitamin C sebanyak 100-200 mgr/hari selama 1 minggu, selanjutnya diberikan 50 mg/hari.
KEKURANGAN ASAM LEMAK ESENSIAL
Asam lemak esensial terdiri dari:
- asam linoleat
- asam linolenat
- asam arakidonat
- asam eikopentaenoat
- asam dokosaheksaenoat.
Asam lemak esensial harus terkandung di dalam makanan sehari-hari.
Di dalam tubuh, asam arakidonat bisa dibuat dari asam linoleat; asam eikosapentanoat dan asam dokosaheksaenoat dapat dibuat dari asam linolenat.
Asam linoleat dan asam linolenat bisa ditemukan dalam minyak sayur (misalnya minyak jagung, minyak biji kapas dan minyak kadang kedele); sedangkan asam eikosapentanoat dan asam dokosaheksaenoat ditemukan dalam minyak ikan.
Asam lemak esensial penting untuk berbagai proses fisiologis, termasuk mempertahankan keutuhan kulit dan struktur selaput sel serta mensintesa senyawa biologis aktif yang penting (misalnya prostaglandin dan leukotrien).
Beberapa penelitian menyebutkan bahwa asam lemak esensial penting untuk perkembangan penglihatan yang normal pada bayi.
Kekurangan asam linoleat bisa terjadi pada bayi yang susu formulanya mengandung sedikit asam lemak tak jenuh ganda.
Gejalanya berupa kulit kering dan bersisik yang kemudian akan mengelupas. Dari lipatan kulit (terutama di sekitar anus) keluar cairan yang menyerupai nanah.
Kekurangan asam lemak esensial juga dapat menyebabkan perubahan yang berarti pada proses metabolisme, yang mempengaruhi kandungan lemak dalam darah, fungsi trombosit, respon peradangan dan respon kekebalan tertentu.
Gejala yang sama dapat ditemukan pada pasen yang menerima makanan melalui infus dalam jangka panjang tetapi tidak mendapatkan asam lemak esensial.
Jika asam linolenat tidak terkandung dalam makanan yang diberikan melalui infus jangka panjang, maka akan terjadi komplikasi neurologis berupa mati rasa, kelemahan, tidak dapat berjalan, nyeri tungkai dan pandangan kabur, disertai kadar asam linolenat yang sangat rendah dalam darah.
Gejala-gejala tersebut akan menghilang setelah diberikan asam linolenat.
Kelainan Metabolisme
Metabolisme adalah proses pengolahan
(pembentukan dan penguraian) zat -zat yang diperlukan oleh tubuh agar
tubuh dapat menjalankan fungsinya.
Kelainan metabolisme seringkali disebabkan oleh kelainan genetik yang mengakibatkan hilangnya enzim tertentu yang diperlukan untuk merangsang suatu proses metabolisme.
KELAINAN METABOLISME KARBOHIDRAT
Karbohidrat adalah gula, diantaranya adalah glukosa, sukrosa dan fruktosa.
Beberapa gula (misalnya sukrosa) harus diproses oleh enzim di dalam tubuh sebelum bisa digunakan sebagai sumber energi. Jika enzim yang diperlukan tidak ada, maka gula akan tertimbun dan menimbulkan masalah kesehatan.
Galaktosemia
Galaktosemia (kadar galaktosa yang tinggi dalam darah) biasanya disebabkan oleh kekurangan enzim galaktose 1-fosfat uridil transferase. Kelainan ini merupakan kelainan bawaan.
Sekitar 1 dari 50.000-70.000 bayi terlahir tanpa enzim tersebut. Pada awalnya mereka tampak normal, tetapi beberapa hari atau beberapa minggu kemudian, nafsu makannya akan berkurang, muntah, tampak kuning (jaundice) dan pertumbuhannya yang normal terhenti.
Hati membesar, di dalam air kemihnya ditemukan sejumlah besar protein dan asam amino, terjadi pembengkakan jaringan dan penimbunan cairan dalam tubuh.
Jika pengobatan tertunda, anak akan memiliki tubuh yang pendek dan mengalami keterbelakangan mental. Banyak yang menderita katarak.
Kebanyakan penyebabnya tidak diketahui.
Diduga suatu galaktosemia jika pada pemeriksaan laboratorium, di dalam air kemih ditemukan galaktosa dan galaktose 1-fostate. Untuk memperkuat diagnosis, dilakukan pemeriksaan darah dan sel-sel hati, yang akan menunjukkan tidak adanya enzim galaktose 1-fosfat uridil transferase.
Susu dan hasil olahan susu (yang merupakan sumber dari galaktosa) tidak boleh diberikan kepada anak yang menderita galaktosemia. Demikian juga halnya dengan beberapa jenis buah-buahan, sayuran dan hasil laut (misalnya rumput laut).
Seorang wanita yang diketahui membawa gen untuk penyakit ini sebaiknya tidak mengkonsumsi galaktosa selama kehamilan.
Seorang wanita hamil yang menderita galaktosemia juga harus menghindari galaktosa. Jika kadar galaktosanya tinggi, galaktosa dapat melewati plasenta dan sampai ke janin, menyebabkan katarak.
Penderita galaktosemia harus menghindari galaktosa seumur hidupnya.
Jika diobati secara adekuat, tidak akan terjadi keterbelakangan mental. Tetapi tingkat kecerdasannya lebih rendah dibandingkan dengan saudara kandungnya dan sering ditemukan gangguan berbicara.
Pada masa pubertas dan masa dewasa, anak perempuan seringkali mengalami kegagalan ovulasi (pelepasan sel telur) dan hanya sedikit yang dapat hamil secara alami.
Glikogenosis
Glikogenosis (Penyakit penimbunan glikogen) adalah sekumpulan penyakit keturunan yang disebabkan oleh tidak adanya 1 atau beberapa enzim yang diperlukan untuk mengubah gula menjadi glikogen atau mengubah glikogen menjadi glukosa (untuk digunakan sebagai energi).
Pada glikogenosis, sejenis atau sejumlah glikogen yang abnormal diendapkan di dalam jaringan tubuh, terutama di hati.
Gejalanya timbul sebagai akibat dari penimbunan glikogen atau hasil pemecahan glikogen atau akibat dari ketidakmampuan untuk menghasilkan glukosa yang diperlukan oleh tubuh.
Usia ketika timbulnya gejala dan beratnya gejala bervariasi, tergantung kepada enzim apa yang tidak ditemukan.
Diagnosis ditegakkan berdasarkan hasil pemeriksaan terhadap contoh jaringan (biasanya otot atau hati), yang menunjukkan adanya enzim yang hilang.
Pengobatan tergantung kepada jenis penyakitnya.
Untuk membantu mencegah turunnya kadar gula darah, dianjurkan untuk mengkonsumsi makanan kaya karbohidrat dalam porsi kecil sebanyak beberapa kali dalam sehari. Pada beberapa anak yang masih kecil, masalah ini bisa diatasi dengan memberikan tepung jagung yang tidak dimasak setiap 4-6 jam. Kadang pada malam hari diberikan larutan karbohidrat melalui selang yang dimasukkan ke lambung.
Penyakit penimbunan glikogen cenderung menyebabkan penimbunan asam urat, yang dapat menyebabkan gout dan batu ginjal. Untuk mencegah hal tersebut seringkali perlu diberikan obat-obatan.
Pada beberapa jenis glikogenesis, untuk mengurangi kram otot, aktivitas anak harus dibatasi.
Jenis & Karakteristik Penyakit Penimbunan Glikogen
Intoleransi Fruktosa Herediter
Intoleransi Fruktosa Herediter adalah suatu penyakit keturunan dimana tubuh tidak dapat menggunakan fruktosa karena tidak memiliki enzim fosfofruktaldolase.
Sebagai akibatnya, fruktose 1-fosfatase (yang merupakan hasil pemecahan dari fruktosa) tertimbun di dalam tubuh, menghalangi pembentukan glikogen dan menghalangi perubahan glikogen menjadi glukosa sebagai sumber energi.
Mencerna fruktosa atau sukrosa (yang dalam tubuh akan diuraikan menjadi fruktosa, kedua jenis gula ini terkandung dalam gula meja) dalam jumlah yang lebih, bisa menyebabkan:
- hipoglikemia (kadar gula darah yang rendah) disertai keringat dingin
- tremor (gerakan gemetar diluar kesadaran)
- linglung
- mual
- muntah
- nyeri perut
- kejang (kadang-kadang)
- koma.
Jika penderita terus mengkonsumsi fruktosa, bisa terjadi kerusakan ginjal dan hati serta kemunduran mental.
Diagnosis ditegakkan berdasarkan hasil pemeriksaan contoh jaringan hati yang menunjukkan adanya enzim yang hilang.
Juga dilakukan pengujian respon tubuh terhadap fruktosa dan glukosa yang diberikan melalui infus.
Karier (pembawa gen untuk penyakit ini tetapi tidak menderita penyakit ini) dapat ditentukan melalui analisa DNA dan membandingkannya dengan DNA penderita dan DNA orang normal.
Pengobatan terdiri dari menghindari fruktosa (biasanya ditemukan dalam buah-buahan yang manis), sukrosa dan sorbitol (pengganti gula) dalam makanan sehari-hari.
Serangan hipoglikemia diatasi dengan pemberian tablet glukosa, yang harus selalu dibawa oleh setiap penderita intoleransi fruktosa herediter.
Fruktosuria
Fruktosuria merupakan suatu keadaan yang tidak berbahaya, dimana fruktosa dibuang ke dalam air kemih.
Fruktosuria disebabkan oleh kekurangan enzim fruktokinase yang sifatnya diturunkan.
1 dari 130.000 penduduk menderita fruktosuria.
Fruktosuria tidak menimbulkan gejala, tetapi kadar fruktosa yang tinggi di dalam darah dan air kemih dapat menyebabkan kekeliruan diagnosis dengan diabetes mellitus.
Tidak perlu dilakukan pengobatan khusus.
Pentosuria
Pentosuria adalah suatu keadaan yang tidak berbahaya, yang ditandai dengan ditemukannya gula xylulosa di dalam air kemih karena tubuh tidak memiliki enzim yang diperlukan untuk mengolah xylulosa.
Pentosuria hampir selalu hanya ditemukan pada orang Yahudi.
Pentosuria tidak menimbulkan masalah kesehatan, tetapi adanya xylulosa dalam air kemih bisa menyebabkan kekeliruan diagnosis dengan diabetes mellitus.
Tidak perlu dilakukan pengobatan khusus.
KELAINAN METABOLISME PIRUVAT
Piruvat terbentuk dalam proses pengolahan karbohidrat, lemak dan protein.
Piruvat merupakan sumber energi untuk mitokondria (komponen sel yang menghasilkan energi).
Gangguan pada metabolisme piruvat dapat menyebabkan terganggunya fungsi mitokondria sehingga timbul sejumlah gejala:
- kerusakan otot
- keterbelakangan mental
- kejang
- penimbunan asam laktat yang menyebabkan asidosis (meningkatnya asam dalam tubuh)
- kegagalan fungsi organ (jantung, paru-paru, ginjal atau hati).
Gejala tersebut dapat terjadi kapan saja, mulai dari awal masa bayi sampai masa dewasa akhir.
Olah raga, infeksi atau alkohol dapat memperburuk gejala, sehingga terjadi asidosis laktat yang berat (kram dan kelemahan otot).
Kekurangan kompleks piruvat dehidrogenase
Kompleks piruvat dehidrogenase adalah sekumpulan enzim yang diperlukan untuk mengolah piruvat.
Kekurangan kompleks piruvat dehidrogenase bisa menyebabkan berkurangnya kadar asetil koenzim A yang penting untuk pembentukan energi.
Gejala utamanya adalah:
- aksi otot menjadi lambat
- koordinasi buruk
- gangguan keseimbangan yang berat yang menyebabkan penderita tidak dapat berjalan.
Gejala lainnya adalah kejang, keterbelakangan mental dan kelainan bentuk otak.
Penyakit ini tidak dapat disembuhkan, tetapi diet tinggi lemak bisa membantu beberapa penderita.
Tidak memiliki piruvat karboksilase
Tidak memiliki enzim piruvat karboksilase akan mempengaruhi atau menghalangi pembentukan glukosa di dalam tubuh. Akibatnya di dalam darah akan tertimbun asam laktat dan keton yang menyebabkan timbulnya mual dan muntah.
Penyakit ini sering berakibat fatal.
Sintesa asam amino (komponen pembentuk protein) juga tergantung kepada piruvat karboksilase.
Jika enzim ini tidak ada, maka pembentukan neurotransmiter (zat yang menghantarkan gelombang saraf) akan berkurang, menyebabkan sejumlah kelainan saraf, termasuk keterbelakangan mental yang berat.
Hipoglikemia (kadar gula darah yang rendah) dan asidosis (penimbunan asam di dalam darah) dapat dikurangi dengan cara sering memakan makanan kaya karbohidrat. Tetapi belum ditemukan obat yang dapat menggantikan neurotransmiter yang hilang.
Pada penyakit yang lebih ringan, bisa dilakukan pembatasan asupan protein.
KELAINAN METABOLISME ASAM AMINO
Asam amino merupakan komponen pembentuk protein.
Penyakit keturunan pada pengolahan asam amino dapat menyebabkan gangguan pada penguraian asam amino maupun pemindahan asam amino ke dalam sel.
Fenilketonuria
Fenilketonuria (Fenilalaninemia, Fenilpiruvat oligofrenia) adalah suatu penyakit keturunan dimana tubuh tidak memiliki enzim pengolah asam amino fenilalanin, sehingga menyebabkan kadar fenilalanin yang tinggi di dalam darah, yang berbahaya bagi tubuh.
Dalam keadaan normal, fenilalanin diubah menjadi tirosin dan dibuang dari tubuh. Tanpa enzim tersebut, fenilalanin akan tertimbun di dalam darah dan merupakan racun bagi otak, menyebabkan keterbelakangan mental.
Pada saat bayi baru lahir biasanya tidak ditemukan gejala. Kadang bayi tampak mengantuk atau tidak mau makan.
Bayi cenderung memiliki kulit, rambut dan mata yang berwarna lebih terang dibandingkan dengan anggota keluarga lainnya yang tidak menderita penyakit ini.
Beberapa bayi mengalami ruam kulit yang menyerupai eksim.
Jika tidak diobati, bayi akan segera mengalami keterbelakangan mental, yang sifatnya biasanya berat.
Gejala pada anak-anak yang menderita fenilketonuria yang tidak diobati atau tidak terdiagnosis adalah:
- kejang
- mual dan muntah
- perilaku agresif atau melukai diri sendiri
- hiperaktif
- gejala psikis (kadang-kadang).
Bau badannya menyerupai tikus karena di dalam air kemih dan keringatnya mengandung asam fenil asetat (hasil pemecahan fenilalanin).
Fenilketonuria pada wanita hamil memberikan dampak yang besar terhadap janin yang dikandungnya, yaitu menyebabkan keterbelakangan mental dan fisik.
Bayi terlahir dengan kepala yang kecil (mikrosefalus) dan penyakit jantung.
Jika selama hamil dilakukan pengawasan ketat terhadap kadar fenilalanin pada ibu, biasanya bayi yang lahir akan normal.
Diagnosis ditegakkan berdasarkan tinginya kadar fenilalanin dan rendahnya kadar tirosin.
Pengobatan meliputi pembatasan asupan fenilalanin.
Semua sumber protein alami mengandung 4% fenilalanin, karena itu mustahil untuk mengkonsumsi protein dalam jumlah yang cukup tanpa melebihi jumlah fenilalanin yang dapat diterima. Karena itu sebagai pengganti susu dan daging, penderita harus makan sejumlah makanan sintetis yang menyediakan asam amino lainnya.
Penderita boleh memakan makanan alami rendah protein, seperti buah-buahan, sayur-sayuran dan gandum dalam jumlah tertentu.
Untuk mencegah terjadinya keterbelakangan mental, pada minggu pertama kehidupan bayi, asupan fenilalanin harus dibatasi. Pembatasan yang dimulai sedini mungkin dan terlaksana dengan baik, memungkinkan terjadinya perkembangan yang normal dan mencegah kerusakan otak. Jika pembatasan ini tidak dapat dipertahankan, maka anak akan mengalami kesulitan di sekolah.
Pembatasan yang dimulai setelah anak berumur 2-3 tahun hanya bisa mengendalikan hiperaktivitas yang berat dan kejang.
Pembatasan asupan fenilalanin sebaiknya dilakukan sepanjang hidup penderita.
Kelainan metabolisme seringkali disebabkan oleh kelainan genetik yang mengakibatkan hilangnya enzim tertentu yang diperlukan untuk merangsang suatu proses metabolisme.
KELAINAN METABOLISME KARBOHIDRAT
Karbohidrat adalah gula, diantaranya adalah glukosa, sukrosa dan fruktosa.
Beberapa gula (misalnya sukrosa) harus diproses oleh enzim di dalam tubuh sebelum bisa digunakan sebagai sumber energi. Jika enzim yang diperlukan tidak ada, maka gula akan tertimbun dan menimbulkan masalah kesehatan.
Galaktosemia
Galaktosemia (kadar galaktosa yang tinggi dalam darah) biasanya disebabkan oleh kekurangan enzim galaktose 1-fosfat uridil transferase. Kelainan ini merupakan kelainan bawaan.
Sekitar 1 dari 50.000-70.000 bayi terlahir tanpa enzim tersebut. Pada awalnya mereka tampak normal, tetapi beberapa hari atau beberapa minggu kemudian, nafsu makannya akan berkurang, muntah, tampak kuning (jaundice) dan pertumbuhannya yang normal terhenti.
Hati membesar, di dalam air kemihnya ditemukan sejumlah besar protein dan asam amino, terjadi pembengkakan jaringan dan penimbunan cairan dalam tubuh.
Jika pengobatan tertunda, anak akan memiliki tubuh yang pendek dan mengalami keterbelakangan mental. Banyak yang menderita katarak.
Kebanyakan penyebabnya tidak diketahui.
Diduga suatu galaktosemia jika pada pemeriksaan laboratorium, di dalam air kemih ditemukan galaktosa dan galaktose 1-fostate. Untuk memperkuat diagnosis, dilakukan pemeriksaan darah dan sel-sel hati, yang akan menunjukkan tidak adanya enzim galaktose 1-fosfat uridil transferase.
Susu dan hasil olahan susu (yang merupakan sumber dari galaktosa) tidak boleh diberikan kepada anak yang menderita galaktosemia. Demikian juga halnya dengan beberapa jenis buah-buahan, sayuran dan hasil laut (misalnya rumput laut).
Seorang wanita yang diketahui membawa gen untuk penyakit ini sebaiknya tidak mengkonsumsi galaktosa selama kehamilan.
Seorang wanita hamil yang menderita galaktosemia juga harus menghindari galaktosa. Jika kadar galaktosanya tinggi, galaktosa dapat melewati plasenta dan sampai ke janin, menyebabkan katarak.
Penderita galaktosemia harus menghindari galaktosa seumur hidupnya.
Jika diobati secara adekuat, tidak akan terjadi keterbelakangan mental. Tetapi tingkat kecerdasannya lebih rendah dibandingkan dengan saudara kandungnya dan sering ditemukan gangguan berbicara.
Pada masa pubertas dan masa dewasa, anak perempuan seringkali mengalami kegagalan ovulasi (pelepasan sel telur) dan hanya sedikit yang dapat hamil secara alami.
Glikogenosis
Glikogenosis (Penyakit penimbunan glikogen) adalah sekumpulan penyakit keturunan yang disebabkan oleh tidak adanya 1 atau beberapa enzim yang diperlukan untuk mengubah gula menjadi glikogen atau mengubah glikogen menjadi glukosa (untuk digunakan sebagai energi).
Pada glikogenosis, sejenis atau sejumlah glikogen yang abnormal diendapkan di dalam jaringan tubuh, terutama di hati.
Gejalanya timbul sebagai akibat dari penimbunan glikogen atau hasil pemecahan glikogen atau akibat dari ketidakmampuan untuk menghasilkan glukosa yang diperlukan oleh tubuh.
Usia ketika timbulnya gejala dan beratnya gejala bervariasi, tergantung kepada enzim apa yang tidak ditemukan.
Diagnosis ditegakkan berdasarkan hasil pemeriksaan terhadap contoh jaringan (biasanya otot atau hati), yang menunjukkan adanya enzim yang hilang.
Pengobatan tergantung kepada jenis penyakitnya.
Untuk membantu mencegah turunnya kadar gula darah, dianjurkan untuk mengkonsumsi makanan kaya karbohidrat dalam porsi kecil sebanyak beberapa kali dalam sehari. Pada beberapa anak yang masih kecil, masalah ini bisa diatasi dengan memberikan tepung jagung yang tidak dimasak setiap 4-6 jam. Kadang pada malam hari diberikan larutan karbohidrat melalui selang yang dimasukkan ke lambung.
Penyakit penimbunan glikogen cenderung menyebabkan penimbunan asam urat, yang dapat menyebabkan gout dan batu ginjal. Untuk mencegah hal tersebut seringkali perlu diberikan obat-obatan.
Pada beberapa jenis glikogenesis, untuk mengurangi kram otot, aktivitas anak harus dibatasi.
Jenis & Karakteristik Penyakit Penimbunan Glikogen
|
Nama
|
Organ yg terkena
|
Gejala
|
Enzim yg hilang
|
|
Tipe O
|
Hati, otot
|
Pembesaran hati disertai penimbunan lemak di dalam sel hati (perlemakan hati)
|
Glikogen sintetase
|
|
Penyakit von Gierke (Tipe IA)
|
Hati, ginjal
|
Pembesaran hati & ginjal
|
Glukose-6-fosfatase
|
|
Sel tipe IB
|
Hati, sel darah putih
|
|
Glukose-6-fosfatase translokase
|
|
Penyakit Pompe (Tipe II)
|
Semua organ
|
Pembesaran hati & jantung
|
Lisosomal glukosidase
|
|
Penyakit Forbes (Tipe III)
|
Hati, otot, jantung, sel darah putih
|
|
Sistem enzim debrancher
|
|
Penyakit Andersen (Tipe IV)
|
Hati, otot, jaringan lainnya
|
|
Sistem enzim brancher
|
|
Penyakit McArdie (Tipe V)
|
Otot
|
Kram otot selama melakukan kegiatan/aktivitas
|
Fosforilase otot
|
|
Penyakit Hers (Tipe VI)
|
Hati
|
|
Fosforilase hati
|
|
Penyakit Tarui (Tipe VII)
|
Otot kerangka, sel darah merah
|
|
Fosfofruktokinase
|
Intoleransi Fruktosa Herediter
Intoleransi Fruktosa Herediter adalah suatu penyakit keturunan dimana tubuh tidak dapat menggunakan fruktosa karena tidak memiliki enzim fosfofruktaldolase.
Sebagai akibatnya, fruktose 1-fosfatase (yang merupakan hasil pemecahan dari fruktosa) tertimbun di dalam tubuh, menghalangi pembentukan glikogen dan menghalangi perubahan glikogen menjadi glukosa sebagai sumber energi.
Mencerna fruktosa atau sukrosa (yang dalam tubuh akan diuraikan menjadi fruktosa, kedua jenis gula ini terkandung dalam gula meja) dalam jumlah yang lebih, bisa menyebabkan:
- hipoglikemia (kadar gula darah yang rendah) disertai keringat dingin
- tremor (gerakan gemetar diluar kesadaran)
- linglung
- mual
- muntah
- nyeri perut
- kejang (kadang-kadang)
- koma.
Jika penderita terus mengkonsumsi fruktosa, bisa terjadi kerusakan ginjal dan hati serta kemunduran mental.
Diagnosis ditegakkan berdasarkan hasil pemeriksaan contoh jaringan hati yang menunjukkan adanya enzim yang hilang.
Juga dilakukan pengujian respon tubuh terhadap fruktosa dan glukosa yang diberikan melalui infus.
Karier (pembawa gen untuk penyakit ini tetapi tidak menderita penyakit ini) dapat ditentukan melalui analisa DNA dan membandingkannya dengan DNA penderita dan DNA orang normal.
Pengobatan terdiri dari menghindari fruktosa (biasanya ditemukan dalam buah-buahan yang manis), sukrosa dan sorbitol (pengganti gula) dalam makanan sehari-hari.
Serangan hipoglikemia diatasi dengan pemberian tablet glukosa, yang harus selalu dibawa oleh setiap penderita intoleransi fruktosa herediter.
Fruktosuria
Fruktosuria merupakan suatu keadaan yang tidak berbahaya, dimana fruktosa dibuang ke dalam air kemih.
Fruktosuria disebabkan oleh kekurangan enzim fruktokinase yang sifatnya diturunkan.
1 dari 130.000 penduduk menderita fruktosuria.
Fruktosuria tidak menimbulkan gejala, tetapi kadar fruktosa yang tinggi di dalam darah dan air kemih dapat menyebabkan kekeliruan diagnosis dengan diabetes mellitus.
Tidak perlu dilakukan pengobatan khusus.
Pentosuria
Pentosuria adalah suatu keadaan yang tidak berbahaya, yang ditandai dengan ditemukannya gula xylulosa di dalam air kemih karena tubuh tidak memiliki enzim yang diperlukan untuk mengolah xylulosa.
Pentosuria hampir selalu hanya ditemukan pada orang Yahudi.
Pentosuria tidak menimbulkan masalah kesehatan, tetapi adanya xylulosa dalam air kemih bisa menyebabkan kekeliruan diagnosis dengan diabetes mellitus.
Tidak perlu dilakukan pengobatan khusus.
KELAINAN METABOLISME PIRUVAT
Piruvat terbentuk dalam proses pengolahan karbohidrat, lemak dan protein.
Piruvat merupakan sumber energi untuk mitokondria (komponen sel yang menghasilkan energi).
Gangguan pada metabolisme piruvat dapat menyebabkan terganggunya fungsi mitokondria sehingga timbul sejumlah gejala:
- kerusakan otot
- keterbelakangan mental
- kejang
- penimbunan asam laktat yang menyebabkan asidosis (meningkatnya asam dalam tubuh)
- kegagalan fungsi organ (jantung, paru-paru, ginjal atau hati).
Gejala tersebut dapat terjadi kapan saja, mulai dari awal masa bayi sampai masa dewasa akhir.
Olah raga, infeksi atau alkohol dapat memperburuk gejala, sehingga terjadi asidosis laktat yang berat (kram dan kelemahan otot).
Kekurangan kompleks piruvat dehidrogenase
Kompleks piruvat dehidrogenase adalah sekumpulan enzim yang diperlukan untuk mengolah piruvat.
Kekurangan kompleks piruvat dehidrogenase bisa menyebabkan berkurangnya kadar asetil koenzim A yang penting untuk pembentukan energi.
Gejala utamanya adalah:
- aksi otot menjadi lambat
- koordinasi buruk
- gangguan keseimbangan yang berat yang menyebabkan penderita tidak dapat berjalan.
Gejala lainnya adalah kejang, keterbelakangan mental dan kelainan bentuk otak.
Penyakit ini tidak dapat disembuhkan, tetapi diet tinggi lemak bisa membantu beberapa penderita.
Tidak memiliki piruvat karboksilase
Tidak memiliki enzim piruvat karboksilase akan mempengaruhi atau menghalangi pembentukan glukosa di dalam tubuh. Akibatnya di dalam darah akan tertimbun asam laktat dan keton yang menyebabkan timbulnya mual dan muntah.
Penyakit ini sering berakibat fatal.
Sintesa asam amino (komponen pembentuk protein) juga tergantung kepada piruvat karboksilase.
Jika enzim ini tidak ada, maka pembentukan neurotransmiter (zat yang menghantarkan gelombang saraf) akan berkurang, menyebabkan sejumlah kelainan saraf, termasuk keterbelakangan mental yang berat.
Hipoglikemia (kadar gula darah yang rendah) dan asidosis (penimbunan asam di dalam darah) dapat dikurangi dengan cara sering memakan makanan kaya karbohidrat. Tetapi belum ditemukan obat yang dapat menggantikan neurotransmiter yang hilang.
Pada penyakit yang lebih ringan, bisa dilakukan pembatasan asupan protein.
KELAINAN METABOLISME ASAM AMINO
Asam amino merupakan komponen pembentuk protein.
Penyakit keturunan pada pengolahan asam amino dapat menyebabkan gangguan pada penguraian asam amino maupun pemindahan asam amino ke dalam sel.
Fenilketonuria
Fenilketonuria (Fenilalaninemia, Fenilpiruvat oligofrenia) adalah suatu penyakit keturunan dimana tubuh tidak memiliki enzim pengolah asam amino fenilalanin, sehingga menyebabkan kadar fenilalanin yang tinggi di dalam darah, yang berbahaya bagi tubuh.
Dalam keadaan normal, fenilalanin diubah menjadi tirosin dan dibuang dari tubuh. Tanpa enzim tersebut, fenilalanin akan tertimbun di dalam darah dan merupakan racun bagi otak, menyebabkan keterbelakangan mental.
Pada saat bayi baru lahir biasanya tidak ditemukan gejala. Kadang bayi tampak mengantuk atau tidak mau makan.
Bayi cenderung memiliki kulit, rambut dan mata yang berwarna lebih terang dibandingkan dengan anggota keluarga lainnya yang tidak menderita penyakit ini.
Beberapa bayi mengalami ruam kulit yang menyerupai eksim.
Jika tidak diobati, bayi akan segera mengalami keterbelakangan mental, yang sifatnya biasanya berat.
Gejala pada anak-anak yang menderita fenilketonuria yang tidak diobati atau tidak terdiagnosis adalah:
- kejang
- mual dan muntah
- perilaku agresif atau melukai diri sendiri
- hiperaktif
- gejala psikis (kadang-kadang).
Bau badannya menyerupai tikus karena di dalam air kemih dan keringatnya mengandung asam fenil asetat (hasil pemecahan fenilalanin).
Fenilketonuria pada wanita hamil memberikan dampak yang besar terhadap janin yang dikandungnya, yaitu menyebabkan keterbelakangan mental dan fisik.
Bayi terlahir dengan kepala yang kecil (mikrosefalus) dan penyakit jantung.
Jika selama hamil dilakukan pengawasan ketat terhadap kadar fenilalanin pada ibu, biasanya bayi yang lahir akan normal.
Diagnosis ditegakkan berdasarkan tinginya kadar fenilalanin dan rendahnya kadar tirosin.
Pengobatan meliputi pembatasan asupan fenilalanin.
Semua sumber protein alami mengandung 4% fenilalanin, karena itu mustahil untuk mengkonsumsi protein dalam jumlah yang cukup tanpa melebihi jumlah fenilalanin yang dapat diterima. Karena itu sebagai pengganti susu dan daging, penderita harus makan sejumlah makanan sintetis yang menyediakan asam amino lainnya.
Penderita boleh memakan makanan alami rendah protein, seperti buah-buahan, sayur-sayuran dan gandum dalam jumlah tertentu.
Untuk mencegah terjadinya keterbelakangan mental, pada minggu pertama kehidupan bayi, asupan fenilalanin harus dibatasi. Pembatasan yang dimulai sedini mungkin dan terlaksana dengan baik, memungkinkan terjadinya perkembangan yang normal dan mencegah kerusakan otak. Jika pembatasan ini tidak dapat dipertahankan, maka anak akan mengalami kesulitan di sekolah.
Pembatasan yang dimulai setelah anak berumur 2-3 tahun hanya bisa mengendalikan hiperaktivitas yang berat dan kejang.
Pembatasan asupan fenilalanin sebaiknya dilakukan sepanjang hidup penderita.
Kelainan Mata Bawaan
GLAUKOMA KONGENITAL
Glaukoma Kongenital adalah peningkatan tekanan di dalam bola mata bayi yang baru lahir (biasanya pada kedua mata).
Glaukoma kongenital terjadi akibat adanya gangguan pada perkembangan saluran pembuangan cairan dari mata. Penyakit ini seringkali diturunkan.
Gejalanya berupa:
- mata berair
- peka terhadap cahaya
- mata merah
- kornea tampak kabur
- kornea membesar.
Untuk menegakkan diagnosis glaukoma kongenital perlu dilakukan pemeriksan degan menggunakan oftalmoskop.
Pemeriksaan mata yang biasa dilakukan:
- Pemeriksaan retina
- Pengukuran tekanan intraokuler dengan menggunakan tonometri
- Pemeriksaan lapang pandang
- Pemeriksaan ketajaman penglihatan
- Pemeriksaan refraksi
- Respon refleks pupil
- Pemeriksaan slit lamp.
Jika tidak diobati, bola mata akan membesar dan hampir dapat dipastikan akan terjadi kebutaan.
Pembedahan yang segera dilakukan setelah bayi lahir akan memberikan peluang terbaik untuk menurunkan tekanan di dalam mata dan untuk mempertahankan fungsi penglihatan.
Glaukoma Kongenital adalah peningkatan tekanan di dalam bola mata bayi yang baru lahir (biasanya pada kedua mata).
Glaukoma kongenital terjadi akibat adanya gangguan pada perkembangan saluran pembuangan cairan dari mata. Penyakit ini seringkali diturunkan.
Gejalanya berupa:
- mata berair
- peka terhadap cahaya
- mata merah
- kornea tampak kabur
- kornea membesar.
Untuk menegakkan diagnosis glaukoma kongenital perlu dilakukan pemeriksan degan menggunakan oftalmoskop.
Pemeriksaan mata yang biasa dilakukan:
- Pemeriksaan retina
- Pengukuran tekanan intraokuler dengan menggunakan tonometri
- Pemeriksaan lapang pandang
- Pemeriksaan ketajaman penglihatan
- Pemeriksaan refraksi
- Respon refleks pupil
- Pemeriksaan slit lamp.
Jika tidak diobati, bola mata akan membesar dan hampir dapat dipastikan akan terjadi kebutaan.
Pembedahan yang segera dilakukan setelah bayi lahir akan memberikan peluang terbaik untuk menurunkan tekanan di dalam mata dan untuk mempertahankan fungsi penglihatan.
KATARAK KONGENITAL
Katarak Kongenital adalah kekeruhan pada lensa mata yang ditemukan pada bayi baru lahir.
Katarak kongenital mungkin bisa disebabkan oleh:
- Galaktosemia
- Sindroma kondrodisplasia
- Rubella kongenital
- Sindroma Down (Trisomi 21)
- Sindroma Pierre-Robin
- Katarak kongenital familial
- Sindroma Hallerman-Streiff
- Sindroma serebrohepatorenalis (Sindroma Lowe)
- Trisomi 13
- Sindroma Conradi
- Sindroma displasia ektodermal
- Sindroma Marinesco-Sjogren.
Lensa yang keruh dapat terlihat tanpa bantuan alat khusus dan tampak sebagai warna keputihan pada pada pupil yang seharusnya berwarna hitam.
Bayi gagal menunjukkan kesadaran visual terhadap lingkungan di sekitarnya dan kadang terdapat nistagmus (gerakan mata yang cepat dan tidak biasa).
Untuk menegakkan diagnosis, dilakukan pemeriksaan mata lengkap oleh seorang ahli mata.
Untuk mencari kemungkinan penyebabnya, perlu dilakukan pemeriksan darah dan rontgen.
Katarak harus diangkat sesegera mungkin agar fungsi penglihatan bisa berkembang secara normal. Katarak dibuang melalui pembedahan, yang diikuti dengan pemasangan lensa intraokuler.
Jika penyebabnya diketahui, maka dilakukan pengobatan terhadap penyebab terjadinya katarak kongenital.
Katarak Kongenital adalah kekeruhan pada lensa mata yang ditemukan pada bayi baru lahir.
Katarak kongenital mungkin bisa disebabkan oleh:
- Galaktosemia
- Sindroma kondrodisplasia
- Rubella kongenital
- Sindroma Down (Trisomi 21)
- Sindroma Pierre-Robin
- Katarak kongenital familial
- Sindroma Hallerman-Streiff
- Sindroma serebrohepatorenalis (Sindroma Lowe)
- Trisomi 13
- Sindroma Conradi
- Sindroma displasia ektodermal
- Sindroma Marinesco-Sjogren.
Lensa yang keruh dapat terlihat tanpa bantuan alat khusus dan tampak sebagai warna keputihan pada pada pupil yang seharusnya berwarna hitam.
Bayi gagal menunjukkan kesadaran visual terhadap lingkungan di sekitarnya dan kadang terdapat nistagmus (gerakan mata yang cepat dan tidak biasa).
Untuk menegakkan diagnosis, dilakukan pemeriksaan mata lengkap oleh seorang ahli mata.
Untuk mencari kemungkinan penyebabnya, perlu dilakukan pemeriksan darah dan rontgen.
Katarak harus diangkat sesegera mungkin agar fungsi penglihatan bisa berkembang secara normal. Katarak dibuang melalui pembedahan, yang diikuti dengan pemasangan lensa intraokuler.
Jika penyebabnya diketahui, maka dilakukan pengobatan terhadap penyebab terjadinya katarak kongenital.
Hydrocephalus
Hydrocephalus
adalah akumulasi abnormal cairan cerebrospinal di dalam otak. Cairan
ini sering meningkatkan tekanan sehingga dapat memeras dan merusak otak.
Hydrocephalus terkadang disebut ?air di dalam otak?. (kata "hydrocephalus" berasal dari bahasa Yunani yang artinya "kepala berair".
Hydrocephalus dapat terjadi sebelum lahir atau kapan saja setelah lahir.
Hydrocephalus terkadang disebut ?air di dalam otak?. (kata "hydrocephalus" berasal dari bahasa Yunani yang artinya "kepala berair".
Hydrocephalus dapat terjadi sebelum lahir atau kapan saja setelah lahir.

PENYEBAB
Hydrocephalus dapat berhubungan dengan beberapa sebab termasuk cacat sejak lahir, pendarahan di otak, infeksi, meningitis, tumor, atau cedera kepala. Banyak bentuk dari hydrocephalus adalah hasil dari terhambatnya cairan cerebrospinal di ventrikel (di otak bagian tengah. Pada cacat sejak lahir, kerusakan fisik dari aliran cairan ke ventrikel biasanya menyebabkan hydrocephalus. Hydrocephalus biasanya mendampingi cacat sejak lahir yang disebut spina bifida (meningomyelocele).
GEJALA
Tanda dan gejala hydrocephalus tergantung pada usia penderita.
* Bayi, tanda yan paling nyata dari hydrocephalus adalah besar kepala yang abnormal. Hal ini terjadi karena tekanan luar yang terus menerus pada otak dan temperung kepala dari hydrocephalus sepanjang perkembangan dan pertumbuhan kepala. (Itulah alasannya kepala bayi selalu diukur dengan hati-hati setiap periksa ke dokter).
Gejala hydrocephalus pada bayi yaitu muntah, mengantuk, gelisah, tidak mampu melihat ke atas dan seizures.
* Pada anak yang lebih tua dan orang dewasa, tidak ada pembesaran dari hydrocephalus (karena tulang tengkorak sudah padat dan tidak dapat membesar). Gejala yang terjadi termasuk sakit kepala, mual, muntah dan kadang-kdang pandangan kabur. Bisa menimbulkan masalah pada keseimbangan dan koordinasi, dan perkembangan yang terlambat dalam berjalan dan berbicara pada anak-anak.
Gelisah, sakit kepala, seizures dan perubahan kepribadian seperti tidak mampu berkonsentrasi dan mengingat bisa terjadi. Mengantuk dan pandangan menjadi dua adalah gejala umum perkembangan hydrocephalus.
PENGOBATAN
Pengobatan hydrocephalus meliputi operasi pemasangan pipa untuk memperlancar aliran cairan yang berlebih dan mengurangi tekanan ke otak. Pipa tersebut fleksible, berupa tabung plastik dengan katup satu arah. Pipa dipasang ke dalam sistem ventrikel pada otak untuk membelokkan alian cairan ke bagian lain dari tubuh, sehingga cairan akan mengalir dan diabsorbsi ke dalam aliran darah.
Prognosis penderita hydrocephalus tergantung pada penyebabnya dan waktu diagnosa dan pengobatan. Banyak penderita hydrocephalus anak-anak hidup normal dengan batasan dan kekurangan yang minim. Pada beberapa kasus kerusakan kognitif pada fungsi bahasa dan non- bahasa bisa terjadi. Masalah infeksi karena pemasangan pipa atau tidak berfungsinya alat perlu dilakukan operasi revisi.
Kelainan Kelamin
Kelainan pada alat kelamin luar
(penis, testis, atau klitoris) biasanya dihasilkan dari kadar pada
hormon seks yang tidak normal pada janin sebelum lahir. Congenital
adrenal hyperlapsia (sebuah gangguan metabolisme) dan kromosom yang
tidak normal biasanya menyebabkan kerusakan kelamin.

Seorang anak kemungkinan dilahirkan dengan kelamin yang tidak jelas laki-laki atau perempuan (kelamin rancu, atau intersex state). Kebanyakan anak dengan kelamin rancu adalah pseudohermaphrodites-yang mana, mereka memiliki alat kelamin luar yang rancu namun salah satunya indung telur atau testis (tidak keduanya). Pseudohermaphrodite secara genetik laki-laki atau perempuan.
Kelainan kelamin laki-laki
Alat kelamin laki-laki dan perempuan terbentuk dari jaringan yang sama dalam embrio. Ketika bertindak berdasarkan testosteron tingkat tinggi sebelum lahir (sama pada janin laki-laki normal), kelamin menjadi penis, skrotum, dan penile urethra. Rendah atau tidak adanya tingkat testosteron membuat terbentuknya klitoris, labia majora, dan separate vaginal dan saluran urethral. Testosteron tingkat intermediate menyebabkan kelamin rancu ; kelamin laki-laki memiliki penis dan testikel kecil yang menetap di perut sebagai gantinya masuk kedalam skrotum (undescended testicle, juga dikenal sebagai cryptorchidism), dan kelamin wanita memiliki klitoris yang membesar dan menyatu dengan labia. Penampakan kelamin pada kedua kelamin sangat mirip.
Pseudohermaphroditism pada laki-laki (better termed undervirilized 46,XY intersex), terjadi pada kelamin laki-laki yang memiliki kelamin luar yang terlihat sebagai wanita tetapi memiliki undescended testikel. Pseudohermaphroditism kemungkinan disebabkan oleh kekurangan sebelum lahir pada hormon kelamin laki-laki (androgen), sebuah kemampuan jaringan tubuh untuk bereaksi terhadap androgen, mengarah kepada hormon kelamin wanita (estrogen), atau ketidakmampuan kromosom.
Setelah terbentuk, testis memproduksi lebih banyak androgen tubuh laki-laki. Tidak ada atau tidak terbentuknya testis disebabkan kekurangan androgen.
Kekurangan androgen selama masa kanak-kanak menyebabkan pembentukan kelamin yang tidak sempurna. Anak laki-laki yang terkena mendapatkan suara yang melengking dan otot pembentukan yang lemah untuk seusianya. Penis, testis, dan skrotum tidak terbentuk. Rambut di kelamin dan rambut di ketiak adalah tipis, dan lengan serta kaki panjangnya tidak normal.
Kekurangan androgen bisa diobati dengan testosteron. Testosteron tersebut biasanya diberikan dengan suntikan atau melalui lapisan kulit. Suntikan dan aplikasi kulit menyebabkan beberapa efek samping dibandingkan menggunakan testosteron yang diminum. Testosteron merangsang pertumbuhan, perkembangan kelamin, dan kesuburan.
Kelainan kelamin perempuan
Pseudohermapdroditism wanita (juga disebut virilization atau overvirilized 46, xx intersex) disebabkan oleh penekanan pada hormon laki-laki tingkat tinggi. Penyebab paling umum adalah melebarnya kelenjar adrenal (congenital adrenal hyperlapsia) yang menghasilkan hormon laki-laki secara berlebihan karena sebuah enzim yang hilang. Hormon laki-laki tidak bisa digantikan dengan hormon wanita yang terjadi pada wanita normal. Kadangkala, hormon laki-laki masuk ke plasenta dari darah si ibu ; misalnya, si ibu habis diberikan obat-obatan seperti progesterone untuk mencegah keguguran, (beberapa progesterone dirubah menjadi testosteron hormon laki-laki dengan janin tersebut) atau si ibu mendapatkan hormon yang dihasilkan tumor, meskipun begitu hal ini lebih jarang terjadi.
Pseudohermaphrodite memiliki alat kelamin dalam wanita namun mempunyai klitoris yang melebar yang menyerupai penis kecil.
Jika anak tersebut ditetapkan berkelamin wanita, operasi dilakukan untuk menciptakan kelamin wanita. Operasi ini bisa termasuk pengurangan klitoris, pembentukan atau perbaikan vagina (vaginoplasty), dan perbaikan urethra.
Congenital adrenal hyperlapsia bisa jadi mengancam nyawa karena hal ini bisa menyebabkan ketidaknormalan yang serius pada elektrolit (sodium dan potassium) di dalam darah. Hal ini didiagnosa dengan tes darah dan diobati dengan kortikosteroid.
GEJALA
Pengamatan diagnosa pada seorang anak dengan kelamin rancu termasuk pemeriksaan fisik dan tes darah untuk menganalisa kromosom ( pola kromosom XY adalah laki-laki dan pola kromosom XX adalah perempuan) dan tingkat hormon (hormon pituitary dan hormon seks laki-laki, atau androgens, seperti testosteron). Sinar X dan ultrasonic pada panggul bisa membantu mengidentifikasi alat kelamin dalam. Pengobatan dengan testosteron bisa membantu memperbesar penis sehingga penetapan untuk kelamin laki-laki lebih realistis.
DIAGNOSA
Banyak ahli percaya bahwa kelamin anak harus ditentukan dengan segera. Sebaliknya, ikatan dengan orangtua pada anak tersebut bisa menjadi lebih sulit, dan anak tersebut bisa mengalami gangguan identitas kelamin. Keputusan untuk menentukan kelamin seorang bayi dengan alat kelamin rancu/ganda tergantung pada beberapa faktor. Hal tersebut meliputi seberapa banyak testosteron janin tersebut mengarah ; apa kemampuan fungsi kelamin (sebagai penentu misalnya, dengan berapa banyak jaringan ereksi yang dipunyai bayi) ; dan kemampuan untuk reproduksi. Faktor lingkungan dan psikologi, seperti persepsi orangtua pada kelamin, juga mempengaruhi keputusan tersebut. Operasi untuk memperbaiki kelamin rancu bisa dilakukan kemudian, khususnya jika pengaruh tersebut rumit. Masalah mendasar yang menyebabkan pseudohermaphroditism bisa juga membutuhkan pengobatan.
Seorang anak kemungkinan dilahirkan dengan kelamin yang tidak jelas laki-laki atau perempuan (kelamin rancu, atau intersex state). Kebanyakan anak dengan kelamin rancu adalah pseudohermaphrodites-yang mana, mereka memiliki alat kelamin luar yang rancu namun salah satunya indung telur atau testis (tidak keduanya). Pseudohermaphrodite secara genetik laki-laki atau perempuan.
Kelainan kelamin laki-laki
Alat kelamin laki-laki dan perempuan terbentuk dari jaringan yang sama dalam embrio. Ketika bertindak berdasarkan testosteron tingkat tinggi sebelum lahir (sama pada janin laki-laki normal), kelamin menjadi penis, skrotum, dan penile urethra. Rendah atau tidak adanya tingkat testosteron membuat terbentuknya klitoris, labia majora, dan separate vaginal dan saluran urethral. Testosteron tingkat intermediate menyebabkan kelamin rancu ; kelamin laki-laki memiliki penis dan testikel kecil yang menetap di perut sebagai gantinya masuk kedalam skrotum (undescended testicle, juga dikenal sebagai cryptorchidism), dan kelamin wanita memiliki klitoris yang membesar dan menyatu dengan labia. Penampakan kelamin pada kedua kelamin sangat mirip.
Pseudohermaphroditism pada laki-laki (better termed undervirilized 46,XY intersex), terjadi pada kelamin laki-laki yang memiliki kelamin luar yang terlihat sebagai wanita tetapi memiliki undescended testikel. Pseudohermaphroditism kemungkinan disebabkan oleh kekurangan sebelum lahir pada hormon kelamin laki-laki (androgen), sebuah kemampuan jaringan tubuh untuk bereaksi terhadap androgen, mengarah kepada hormon kelamin wanita (estrogen), atau ketidakmampuan kromosom.
Setelah terbentuk, testis memproduksi lebih banyak androgen tubuh laki-laki. Tidak ada atau tidak terbentuknya testis disebabkan kekurangan androgen.
Kekurangan androgen selama masa kanak-kanak menyebabkan pembentukan kelamin yang tidak sempurna. Anak laki-laki yang terkena mendapatkan suara yang melengking dan otot pembentukan yang lemah untuk seusianya. Penis, testis, dan skrotum tidak terbentuk. Rambut di kelamin dan rambut di ketiak adalah tipis, dan lengan serta kaki panjangnya tidak normal.
Kekurangan androgen bisa diobati dengan testosteron. Testosteron tersebut biasanya diberikan dengan suntikan atau melalui lapisan kulit. Suntikan dan aplikasi kulit menyebabkan beberapa efek samping dibandingkan menggunakan testosteron yang diminum. Testosteron merangsang pertumbuhan, perkembangan kelamin, dan kesuburan.
Kelainan kelamin perempuan
Pseudohermapdroditism wanita (juga disebut virilization atau overvirilized 46, xx intersex) disebabkan oleh penekanan pada hormon laki-laki tingkat tinggi. Penyebab paling umum adalah melebarnya kelenjar adrenal (congenital adrenal hyperlapsia) yang menghasilkan hormon laki-laki secara berlebihan karena sebuah enzim yang hilang. Hormon laki-laki tidak bisa digantikan dengan hormon wanita yang terjadi pada wanita normal. Kadangkala, hormon laki-laki masuk ke plasenta dari darah si ibu ; misalnya, si ibu habis diberikan obat-obatan seperti progesterone untuk mencegah keguguran, (beberapa progesterone dirubah menjadi testosteron hormon laki-laki dengan janin tersebut) atau si ibu mendapatkan hormon yang dihasilkan tumor, meskipun begitu hal ini lebih jarang terjadi.
Pseudohermaphrodite memiliki alat kelamin dalam wanita namun mempunyai klitoris yang melebar yang menyerupai penis kecil.
Jika anak tersebut ditetapkan berkelamin wanita, operasi dilakukan untuk menciptakan kelamin wanita. Operasi ini bisa termasuk pengurangan klitoris, pembentukan atau perbaikan vagina (vaginoplasty), dan perbaikan urethra.
Congenital adrenal hyperlapsia bisa jadi mengancam nyawa karena hal ini bisa menyebabkan ketidaknormalan yang serius pada elektrolit (sodium dan potassium) di dalam darah. Hal ini didiagnosa dengan tes darah dan diobati dengan kortikosteroid.
GEJALA
Pengamatan diagnosa pada seorang anak dengan kelamin rancu termasuk pemeriksaan fisik dan tes darah untuk menganalisa kromosom ( pola kromosom XY adalah laki-laki dan pola kromosom XX adalah perempuan) dan tingkat hormon (hormon pituitary dan hormon seks laki-laki, atau androgens, seperti testosteron). Sinar X dan ultrasonic pada panggul bisa membantu mengidentifikasi alat kelamin dalam. Pengobatan dengan testosteron bisa membantu memperbesar penis sehingga penetapan untuk kelamin laki-laki lebih realistis.
DIAGNOSA
Banyak ahli percaya bahwa kelamin anak harus ditentukan dengan segera. Sebaliknya, ikatan dengan orangtua pada anak tersebut bisa menjadi lebih sulit, dan anak tersebut bisa mengalami gangguan identitas kelamin. Keputusan untuk menentukan kelamin seorang bayi dengan alat kelamin rancu/ganda tergantung pada beberapa faktor. Hal tersebut meliputi seberapa banyak testosteron janin tersebut mengarah ; apa kemampuan fungsi kelamin (sebagai penentu misalnya, dengan berapa banyak jaringan ereksi yang dipunyai bayi) ; dan kemampuan untuk reproduksi. Faktor lingkungan dan psikologi, seperti persepsi orangtua pada kelamin, juga mempengaruhi keputusan tersebut. Operasi untuk memperbaiki kelamin rancu bisa dilakukan kemudian, khususnya jika pengaruh tersebut rumit. Masalah mendasar yang menyebabkan pseudohermaphroditism bisa juga membutuhkan pengobatan.
Kelainan Dinding Abdominal(Omphalocele dan Gastroschisis)


Omphacele biasanya dihubungkan dengan cacat bawaan (seperti cacat jantung) dan dengan sindroma genetika spesifik. Operasi penutupan adalah perawatan terpilih. Namun demikian, kulit dari diding perut harus sering direntangkan sebelum operasi sehingga ada cukup jaringan untuk menutup lubang. Cacat yang luas seringkali membutuhkan penutupan kulit.
Pada gastroschisis, usus besar dapat rusak dengan tekanan dan kontak oleh cairan amoniak. Operasi penutupan adalah perawatan terpilih. Hernia yang besar membutuhkan pembuatan sebuah “silo”, dimana usus besar yang terbuka dibungkus dengan sehelai lapisan pelindung dan digantungkan di atas bayi untuk beberapa hari atau minggu, silo secara perlahan di tekan, memaksa usus kembali ke dalam perut.
Tidak ada komentar:
Posting Komentar